Clear Sky Science · en
Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR
Reprogramming the Failing Heart
Heart failure affects millions of people, often developing slowly after years of high blood pressure or valve disease. In these conditions, heart muscle cells not only grow larger but also switch on a "fetal" genetic program and the heart fills with scar tissue. This study explores a new way to nudge the heart’s own gene-control machinery back toward health—without cutting DNA—by gently turning up a protective regulator called KLF15 in heart muscle cells.
When Heart Cells Lose Their Identity
In a healthy adult heart, cardiomyocytes—heart muscle cells—burn fats efficiently for energy and maintain a stable pattern of gene activity. Using single-cell RNA sequencing in mice exposed to chronic pressure overload, the researchers mapped how individual cardiomyocytes change as the heart moves from normal function to enlargement and finally to failure. They found that a transcription factor called KLF15, which normally keeps metabolism and growth in balance, showed the strongest activity change in diseased cells. As stress increased, KLF15 levels fell and its ability to keep fetal and stress-related genes in check weakened. Similar drops in KLF15 were seen in human hearts from patients with dilated and hypertrophic cardiomyopathy, indicating that this disruption is conserved across species.
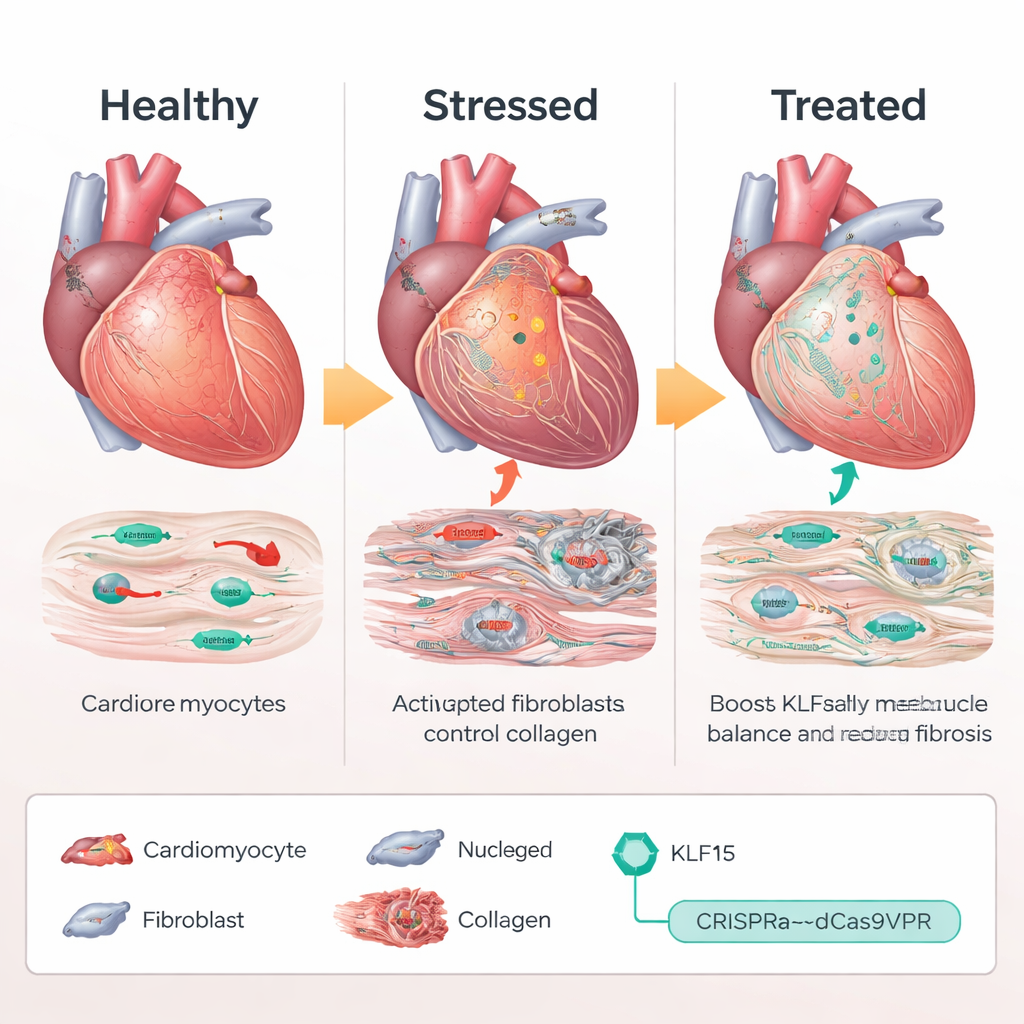
Using CRISPR as a Volume Knob, Not a Pair of Scissors
Rather than adding an extra copy of the KLF15 gene or cutting DNA, the team used a CRISPR-based “activation” system, called dCas9VPR, that binds near the natural Klf15 gene and boosts its own expression. In mice engineered to express this CRISPR activator only in cardiomyocytes, the scientists delivered guide RNAs with an adeno-associated virus (AAV9) to target the Klf15 promoter. Under chronic pressure overload, mice receiving Klf15-activating guides maintained near-normal Klf15 levels. Their heart muscle cells stayed smaller, pumping function declined less, and survival improved compared with control animals. At the molecular level, stress and fetal genes quieted down, while metabolic and calcium-handling genes rebounded, indicating that the unhealthy transcriptional program had been largely reset.
Quieting Scar Formation Through Cell-to-Cell Crosstalk
Heart failure is driven not only by sick muscle cells but also by fibroblasts, support cells that produce collagen and form stiff scar tissue. Single-cell analyses and tissue imaging showed that restoring Klf15 in cardiomyocytes reduced fibroblast activation and overall fibrosis, even though the gene therapy never directly targeted fibroblasts. The team traced this effect to a secreted protein called AZGP1. When Klf15 was boosted in cardiomyocytes, AZGP1 production and release increased. In both mouse hearts and human stem cell–derived heart tissues, higher AZGP1 dampened the TGF-β / SMAD pathway in fibroblasts—a key driver of scarring—lowering levels of markers like α-SMA and POSTN. Importantly, AZGP1 overexpression in cardiomyocytes alone did not reprogram the muscle cells, showing that KLF15 primarily protects cardiomyocytes directly and uses AZGP1 as a messenger to restrain fibroblasts.
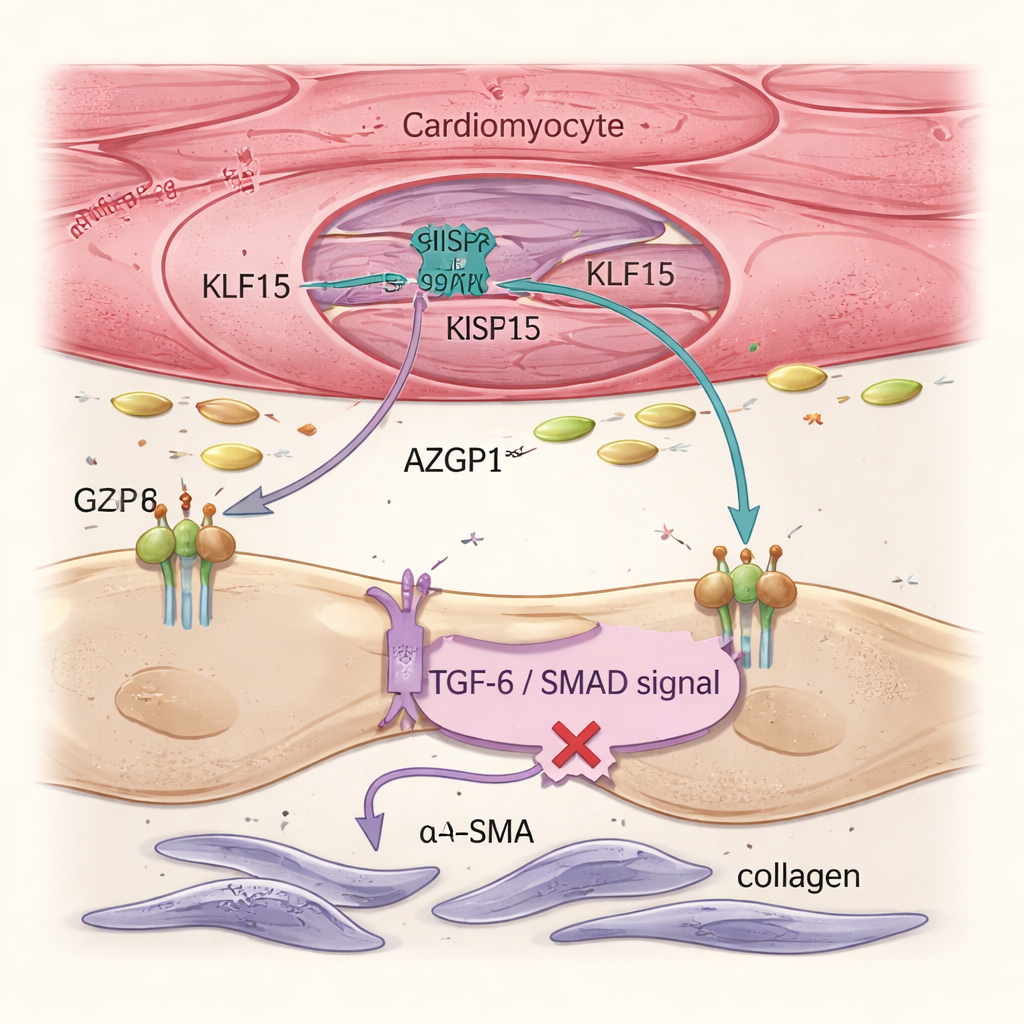
Human Tissue Models Confirm the Protective Circuit
To test whether these mechanisms hold in human cells, the researchers used induced pluripotent stem cell–derived cardiomyocytes grown in three-dimensional engineered heart tissues. When subjected to mechanical load that mimics high blood pressure, these tissues lost KLF15, turned on stress and fetal genes, stiffened, and their contractions weakened—recapitulating disease features. CRISPRa-driven restoration of KLF15 prevented this decline, preserved force generation, and shifted gene expression back toward mature metabolism and structure. Detailed experiments showed that TGF-β1, a well-known pro-fibrotic signal, reduces KLF15 in human cardiomyocytes via its SMAD2/3 pathway, helping explain how chronic stress leads to maladaptive remodeling. Finally, the team engineered a compact, "mini" CRISPRa system based on a smaller Cas9 variant that fits into a single AAV9 vector and is driven by a cardiomyocyte-specific promoter. In precision-cut slices of failing human heart tissue, this vector successfully raised KLF15 levels and improved contractile performance over days in culture.
A Blueprint for Gentler Gene Therapy
For a non-specialist, the core message is that this work shows how carefully turning up a single protective regulator inside heart muscle cells can both stabilize their identity and send signals that limit scarring. By using a CRISPR-based activator that does not cut DNA, the approach fine-tunes the heart’s own gene rather than inserting an artificial one. The study defines a TGF-β → KLF15 → AZGP1 pathway that links mechanical stress to harmful remodeling and demonstrates, in mice, human cell models, and human heart tissue slices, that restoring KLF15 can interrupt this chain reaction. While still at a preclinical stage, the compact, cardiomyocyte-targeted CRISPRa system presented here offers a potential roadmap for treating common, non-genetic forms of heart failure by reprogramming gene activity rather than rewriting the genome.
Citation: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Keywords: heart failure, KLF15, CRISPR activation, cardiac fibrosis, AZGP1