Clear Sky Science · en
Thromboxane receptor activation in dendritic cells mitigates sepsis by suppressing S100a8/a9-mediated neutrophil recruitment
Why taming runaway infections matters
Sepsis is a life-threatening overreaction of the body to infection, often starting in the lungs or gut and leading to multiple organ failure. Even with modern intensive care, sepsis kills millions each year because standard treatments mainly support failing organs rather than precisely calming the misfiring immune system. This study uncovers a previously hidden “brake” inside a key immune cell type—dendritic cells—that can dial back harmful inflammation without shutting down the body’s ability to fight germs.
Immune gatekeepers under pressure
Dendritic cells act as sentinels of the immune system: they sense danger, alert other immune cells, and help decide how strong a response should be. In blood samples from patients with sepsis, the authors found that dendritic cells were not only fewer in number, but also showed sharply reduced levels of a receptor called TP, which normally responds to a fatty molecule named thromboxane A₂. Patients whose dendritic cells had the lowest TP levels tended to have more circulating neutrophils—a first-line infection-fighting white blood cell—and more severe disease, suggesting that when this dendritic-cell brake fails, inflammation can spiral out of control.
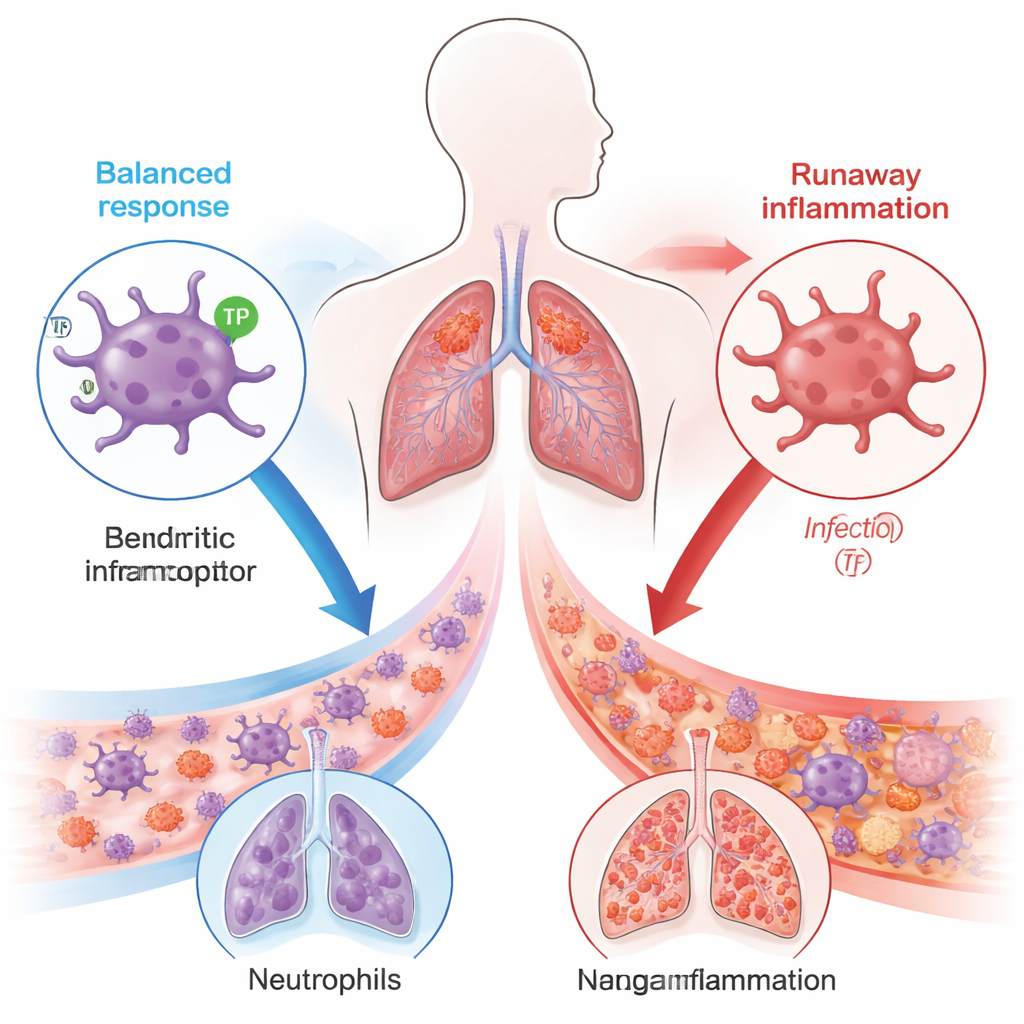
When the brake fails, neutrophils flood the lungs
To probe cause and effect, the team used mouse models of sepsis triggered either by puncturing the intestine (a procedure that releases gut bacteria into the abdomen) or by giving bacterial toxins. Mice engineered to lack TP only in their dendritic cells fared much worse: they died more often, had leakier lungs filled with fluid, and showed signs of heavy neutrophil invasion and tissue damage. When dendritic cells taken from TP-deficient animals were transferred into healthy mice, those recipients also developed more severe sepsis, confirming that faulty signaling in dendritic cells alone can tip the balance toward lethal inflammation.
A danger signal that summons too many defenders
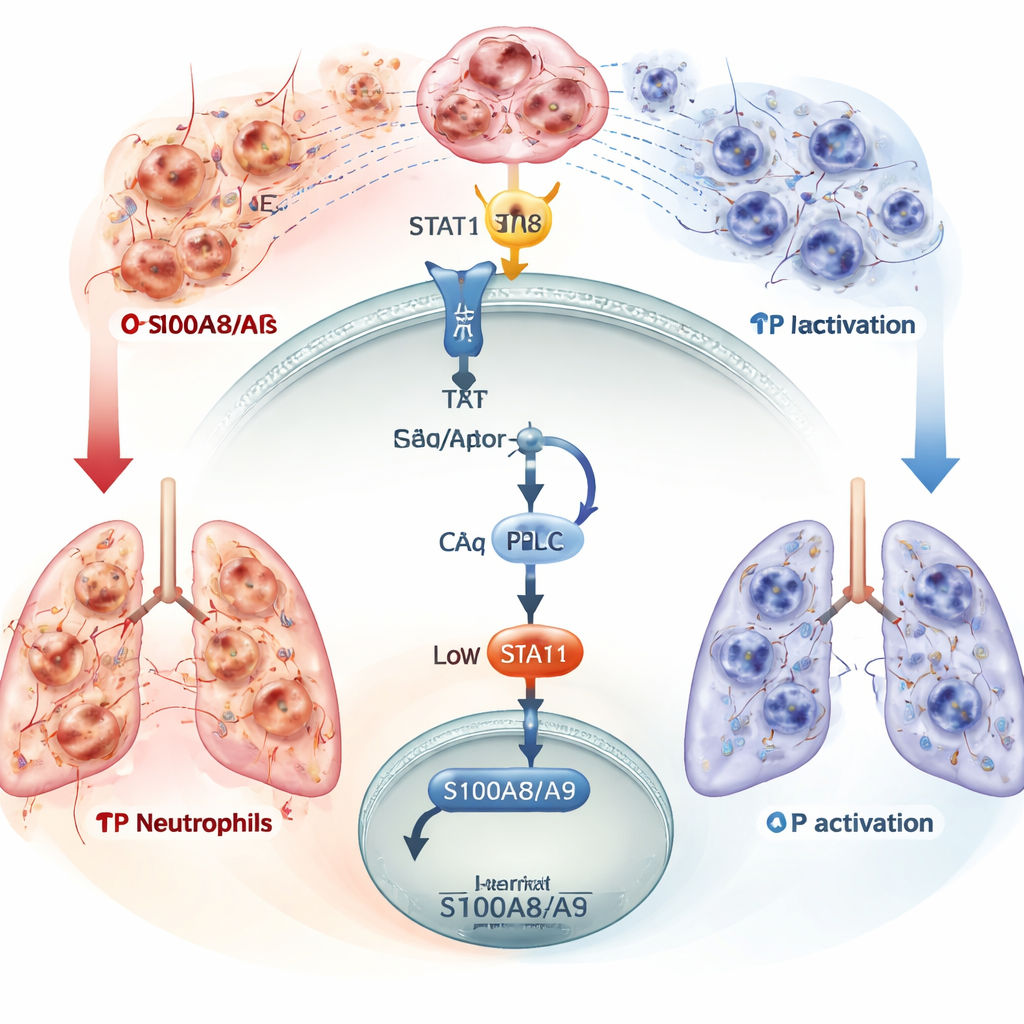
Digging deeper, the researchers examined which genes changed inside dendritic cells when TP was missing. Two danger-associated proteins, S100A8 and S100A9, stood out as strongly increased. These molecules act like flares that call neutrophils into inflamed tissues. The team showed that dendritic cells from septic mice attracted more neutrophils in laboratory tests, and blocking S100A8/A9 with a drug sharply reduced this pull. In both mice and patients, higher S100A8/A9 levels went hand-in-hand with lower TP levels. In live animals, deleting S100A9 specifically in dendritic cells—or blocking its major receptor, the immune sensor TLR4—cut neutrophil influx, reduced the formation of sticky DNA-protein webs called neutrophil extracellular traps (NETs), and protected the lungs from injury.
The signaling circuit behind a targeted brake
The authors then mapped how TP controls S100A8/A9 production inside dendritic cells. Activation of TP triggered an internal chain of events involving a protein kinase (PKCδ) and a transcription factor called STAT1, which moves to the nucleus to influence gene activity. When this pathway was intact, STAT1 helped keep S100A8/A9 levels in check, limiting neutrophil recruitment. Blocking PKCδ or STAT1 broke this protective circuit, allowing S100A8/A9 to surge. Finally, the team built a nano-sized drug that couples a TP-activating compound to a peptide that homes specifically to dendritic cells. In septic mice, this targeted treatment restored the TP signal only in dendritic cells, lowered S100A8/A9, reduced neutrophil and NET buildup in the lungs, and improved survival—all without broadly suppressing the immune system.
Turning a discovery into future sepsis therapies
To a lay reader, the main message is that not all inflammation is bad—but too much, in the wrong place, can be deadly. This work identifies a precise circuit in dendritic cells that normally prevents neutrophils from overwhelming the lungs during severe infection. When TP signaling is lost, dendritic cells overproduce the S100A8/A9 alarm signal, summoning waves of neutrophils that damage tissues instead of helping. By reactivating TP only on dendritic cells—or by blocking the S100A8/A9 pathway—it may be possible to cool the harmful side of inflammation while preserving much of the body’s ability to fight infection. Although still in animal models, this targeted strategy offers a promising direction for future, more precise sepsis treatments.
Citation: Du, R., Pan, T., Wang, Y. et al. Thromboxane receptor activation in dendritic cells mitigates sepsis by suppressing S100a8/a9-mediated neutrophil recruitment. Sig Transduct Target Ther 11, 75 (2026). https://doi.org/10.1038/s41392-026-02592-w
Keywords: sepsis, dendritic cells, neutrophils, inflammation, immune regulation