Clear Sky Science · en
Exogenous Epstein–Barr virus nuclear antigen 1 induces ADAR1-driven tumor resistance against immunotherapy
Why a common virus matters for cancer treatment
Many cancers are now treated with immunotherapy drugs that unleash the body’s own immune system. Yet most patients still do not benefit, because their tumors learn how to hide from immune attack. This study uncovers how a very common virus, Epstein–Barr virus (EBV), helps tumors switch off immune defenses and resist these powerful drugs—and how a new type of designer molecule may switch those defenses back on.
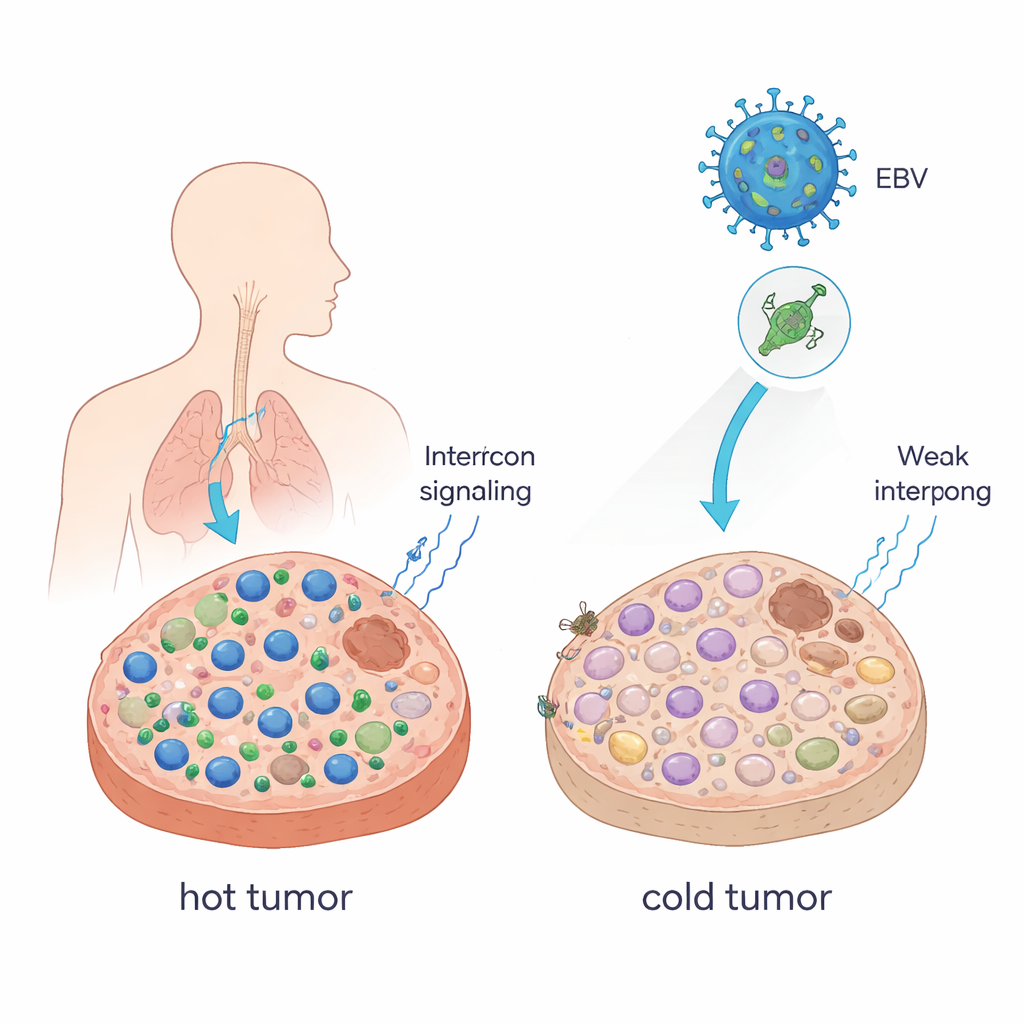
Turning hot tumors into cold ones
Doctors often describe tumors as "hot" when they are full of cancer-killing T cells, and "cold" when those cells are scarce. Hot tumors usually respond well to immune checkpoint blockade (ICB) drugs such as anti–PD-1 antibodies; cold tumors often do not. The authors showed that a single EBV protein, called EBNA1, can push tumors toward this colder, more evasive state. When they forced mouse tumor cells to produce EBNA1 and grew them in mice with intact immune systems, the tumors became larger, contained fewer CD8+ T cells and natural killer cells, and had more immune-suppressing macrophages. Signals called interferons—key messengers that help rally immune cells—were also strongly reduced. In patient samples of nasopharyngeal carcinoma, a cancer tightly linked to EBV, tumors that expressed EBNA1 similarly showed fewer CD8+ T cells than normal tissue.
A viral shortcut into the cell’s RNA-control machinery
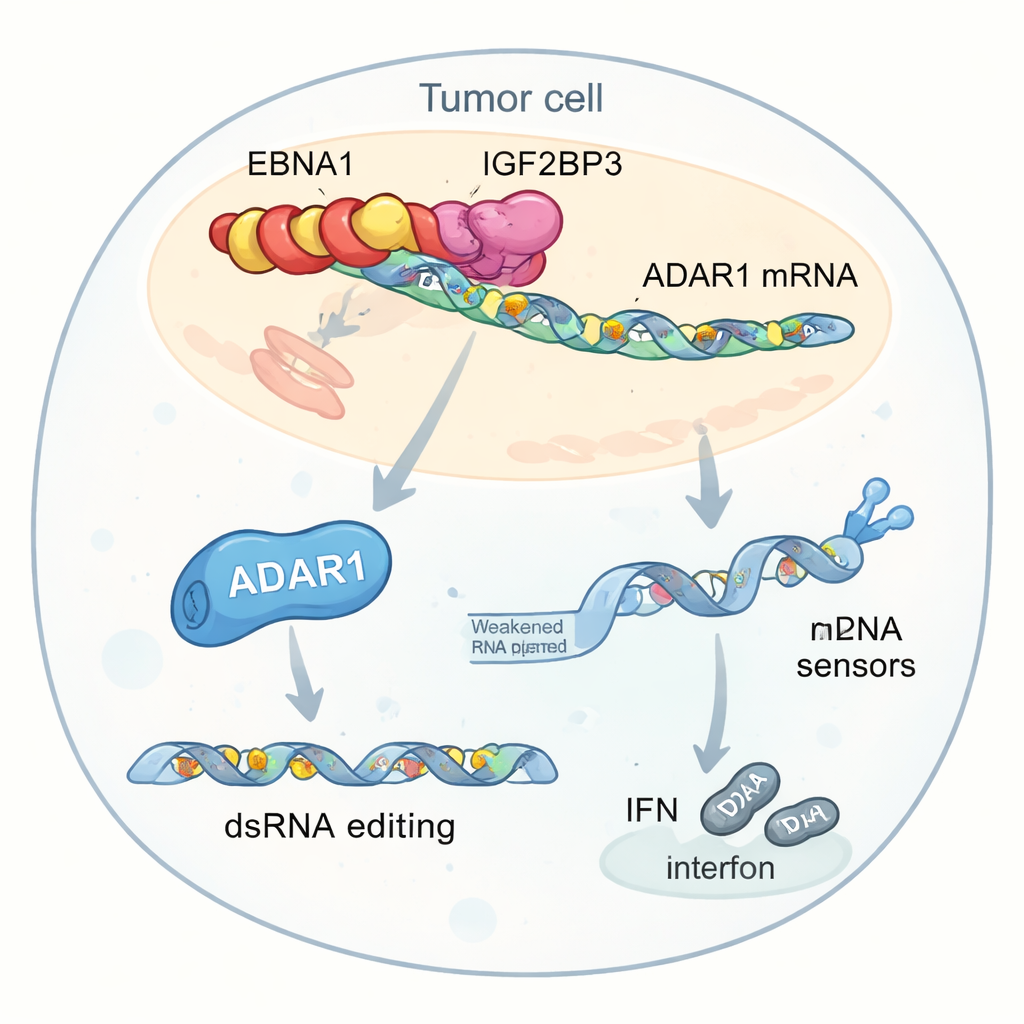
To understand how EBNA1 reshapes the tumor environment, the researchers looked for human proteins that physically interact with it. They focused on a protein called IGF2BP3, which reads small chemical marks (m6A) on messenger RNAs and can boost their stability or translation into protein. EBNA1 bound tightly to IGF2BP3 in several cell types, including EBV-positive cancer cells. Data from patient tumors showed that high IGF2BP3 levels went hand in hand with weak interferon-related gene activity and lower CD8+ T-cell infiltration, hinting that this viral–host alliance could dampen anti-tumor immunity.
Boosting an RNA editor that silences danger signals
Deeper analysis highlighted one key target of this interaction: ADAR1, an enzyme that edits double-stranded RNA by changing certain “A” letters into “I”. This editing can make viral-like RNA inside cells appear less dangerous to internal sensors, reducing interferon responses. The authors found that EBNA1, IGF2BP3 and a translation factor called EIF4G1 form a three-part complex on ADAR1 mRNA. This complex increases m6A tagging, recruits the translation machinery, and selectively boosts ADAR1 protein production without raising its RNA levels. As a result, tumor cells perform more RNA editing in repeated genetic elements located near interferon-related genes. These edits reduce the pool of unedited double-stranded RNA that would normally alert sensors such as MDA5 and PKR, blunting interferon production and helping tumors hide from immune attack.
Less interferon, weaker immunotherapy
When tumor cells expressing EBNA1 were exposed to T cells and anti–PD-1 antibodies in the lab, they were harder to kill than control cells and released less interferon. Even when treated directly with interferon, EBNA1-bearing cells were less sensitive, and their internal RNA sensors were less strongly activated. Reducing the levels of ADAR1 partially reversed these effects, restoring sensor activity and interferon signaling. Genetic and sequencing experiments confirmed that EBNA1-expressing cells showed more A-to-I editing events in specific RNA regions, particularly after interferon stimulation, further supporting the idea that viral boosting of ADAR1 helps neutralize danger signals that would otherwise trigger strong immune responses.
A designer degrader that reawakens immune attack
The team then asked whether stripping tumors of EBNA1 could restore their vulnerability to immunotherapy. They designed a PROTAC molecule, EP-1215, that tags EBNA1 for destruction by the cell’s own disposal system. At low doses, EP-1215 efficiently degraded EBNA1 and reduced ADAR1 protein levels. In mouse experiments, EP-1215 alone had limited impact on EBNA1-positive tumors, and anti–PD-1 alone was also weak. But when combined, the two treatments sharply shrank tumors, increased CD8+ T-cell infiltration, and boosted interferon-producing T cells. In humanized mouse models carrying human immune cells and EBV-related tumors, the combination again outperformed single treatments, without obvious liver or kidney toxicity.
What this means for future cancer care
For non-specialists, the message is that a common virus can quietly rewire cancer cells to dampen internal alarm systems, turning off chemical signals that would otherwise attract and activate immune cells. EBNA1 does this by hijacking a host RNA reader (IGF2BP3) and a translation factor (EIF4G1) to overproduce the RNA editor ADAR1, which edits away the very RNA structures that immune sensors are built to recognize. By degrading EBNA1 with a tailored PROTAC like EP-1215, the authors were able to restore these danger signals and make resistant tumors responsive again to existing checkpoint drugs. If similar strategies prove safe and effective in people, targeting viral helpers such as EBNA1 could offer a new way to convert cold, EBV-linked tumors into hot targets that modern immunotherapies can finally hit.
Citation: Liu, C., Sun, Z., Li, C. et al. Exogenous Epstein–Barr virus nuclear antigen 1 induces ADAR1-driven tumor resistance against immunotherapy. Sig Transduct Target Ther 11, 63 (2026). https://doi.org/10.1038/s41392-026-02574-y
Keywords: Epstein-Barr virus, immunotherapy resistance, ADAR1, RNA editing, nasopharyngeal carcinoma