Clear Sky Science · en
Molecular mechanism of cholesterol-dependent membrane fusion in SARS-CoV-2 entry
Why Cholesterol Matters for a Respiratory Virus
The virus that causes COVID-19, SARS-CoV-2, gets into our cells by fusing its outer coat with our cell membranes. This paper asks a deceptively simple question with big implications: how much does cholesterol—a fatty substance better known from heart disease—shape that fusion step? The authors show that cholesterol in the viral membrane doesn’t just tweak infection efficiency; it actually helps organize the virus’s spike proteins into powerful docking platforms that make entry easier and more reliable.
Building a Minimal Model of Viral Entry
To dissect this process, the researchers rebuilt the virus–cell encounter in a test tube using tiny fat bubbles called liposomes. One set of liposomes carried the SARS-CoV-2 spike protein and stood in for the viral membrane; the other carried the ACE2 receptor, mimicking the host cell surface. When mixed and activated by specific enzymes that “cut” the spike into its fusion-ready form, these artificial membranes fused, allowing a fluorescent dye to move from one bubble to the other. This stripped-down system let the team precisely adjust the lipid composition—including cholesterol levels—on each side and watch how fusion unfolded step by step. 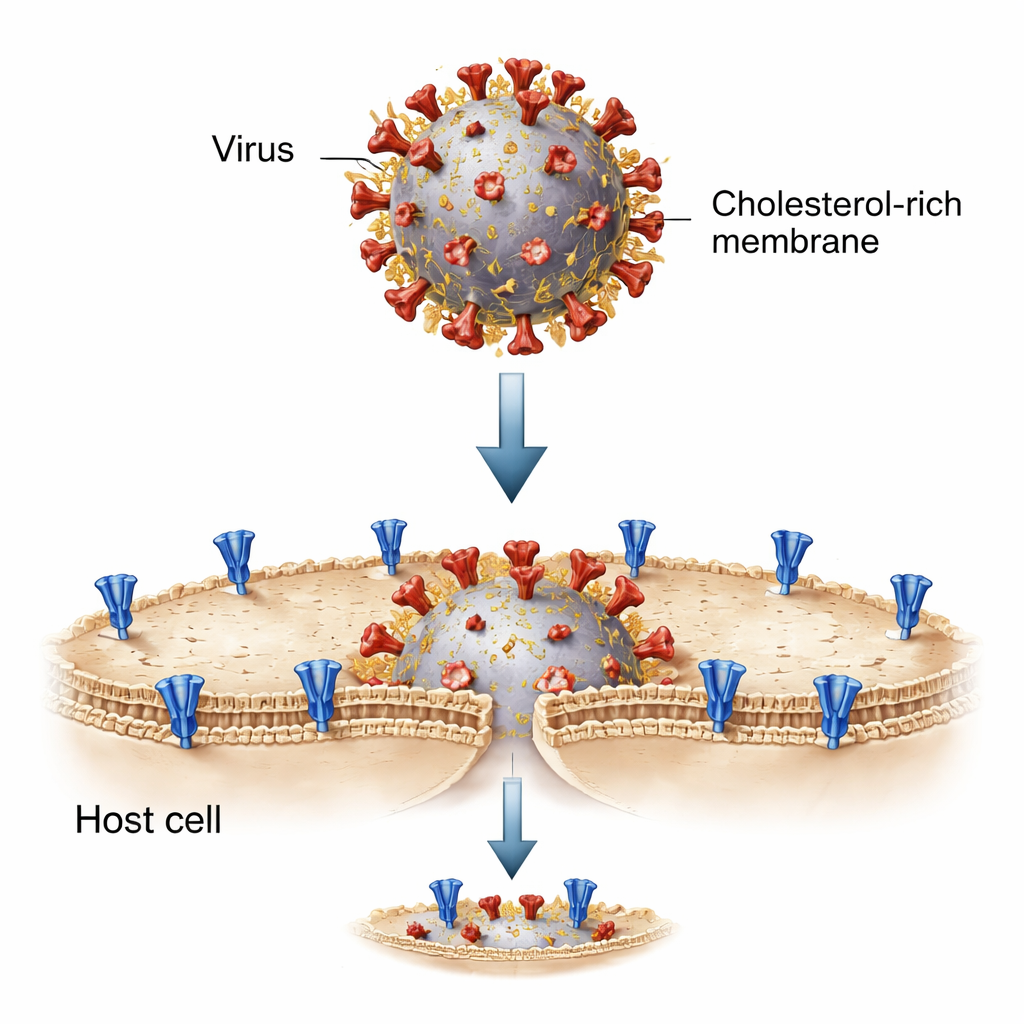
Cholesterol Boosts Docking More Than Fusion
By dialing cholesterol up and down, the team discovered that adding cholesterol to the spike-bearing membrane made fusion more frequent, but not in the way one might expect. High cholesterol did not dramatically change the chance that two already-attached membranes would actually merge. Instead, it sharply increased how often spike-carrying vesicles first docked onto ACE2-bearing vesicles. Single-particle imaging showed many more successful docking events when spike sat in cholesterol-rich membranes, while the probability that a docked pair would go on to fully fuse stayed roughly constant. Interestingly, loading cholesterol into the ACE2 side had little benefit and at very high levels even hindered fusion, pointing to viral—rather than host—cholesterol as the main driver.
From Model Membranes to Living Cells
The researchers then asked whether the same pattern held in living cells. They engineered human cells to express either spike or ACE2 and watched them form large fused structures, or syncytia, when mixed together. Removing cholesterol from spike-expressing cells almost abolished syncytia formation, while restoring cholesterol rescued fusion. In parallel experiments with SARS-CoV-2 “pseudoviruses” (harmless viral particles that use the coronavirus spike to enter cells), stripping cholesterol from the viral membrane sharply reduced infection, and adding it back enhanced infection in a dose-dependent fashion. In contrast, altering cholesterol on ACE2-expressing cells alone produced little change. Across all assays, the message was consistent: cholesterol in the spike-containing membrane is essential for efficient entry.
Spike Clusters: Cholesterol’s Secret Weapon
Why would cholesterol on the viral side matter so much? High-resolution imaging of cell membranes revealed that spike proteins tend to form dense clusters when cholesterol is abundant but remain more scattered when it is depleted. Single-molecule measurements went further, showing that these clusters contain more copies of spike under cholesterol-rich conditions. The authors traced this effect to a cysteine-rich region (CRR) at the tail end of spike, inside the viral membrane. This segment is modified by palmitoylation—a type of fatty “anchor” that favors cholesterol-rich patches. When the team truncated the spike’s tail or mutated all ten cysteines to block palmitoylation, spike no longer clustered with cholesterol, and the cholesterol-driven boost in docking and fusion disappeared. 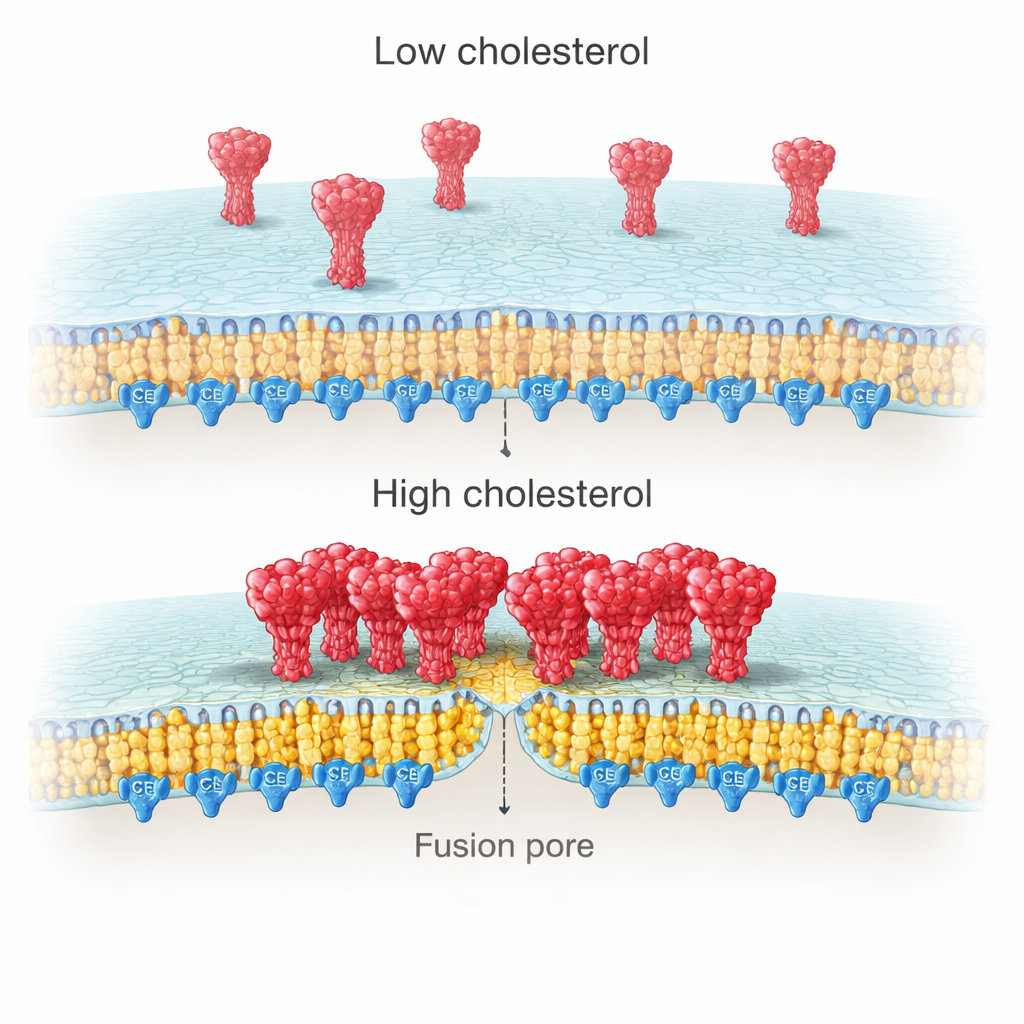
How This Could Help Future Treatments
Put simply, the study concludes that cholesterol turns the viral membrane into a fusion platform by gathering many spike proteins into tight clusters through their palmitoylated tail region. These clusters dock more efficiently onto ACE2 on host cells, increasing the odds that any given encounter will lead to successful fusion and infection. For non-specialists, the key takeaway is that cholesterol is not just a passive ingredient in the viral coat; it is an active organizer of the spike machinery. This makes the spike’s cysteine-rich, cholesterol-sensitive tail—and the enzymes that palmitoylate it—promising targets for broad antiviral strategies that could work across different coronavirus variants.
Citation: Li, W., Wu, M., Feng, S. et al. Molecular mechanism of cholesterol-dependent membrane fusion in SARS-CoV-2 entry. Sig Transduct Target Ther 11, 57 (2026). https://doi.org/10.1038/s41392-026-02573-z
Keywords: cholesterol, SARS-CoV-2 spike, membrane fusion, viral entry, palmitoylation