Clear Sky Science · en
TRIM21-mediated degradation of HILPDA overcomes anti-PD-1 immunotherapy resistance in breast cancer by limiting PD-L1 palmitoylation
Why some cancer immunotherapies stop working
Drugs that unleash the immune system, such as anti–PD‑1 therapies, have transformed treatment for several cancers, including aggressive forms of breast cancer. Yet many tumors either never respond or eventually find ways to escape. This study digs into one such escape route in triple‑negative breast cancer and shows how rewiring tumor fat metabolism—and a repurposed drug called fenretinide—might reopen the door for the immune system when standard immunotherapy fails. 
A hidden helper that shields tumors
The researchers focused on a little‑known protein called HILPDA, previously linked to how cells handle fats under stress. By comparing mouse breast tumors that stayed sensitive to anti‑PD‑1 therapy with tumors that had become resistant, they found that HILPDA levels were much higher in the resistant cancers. Analyses of large patient databases and tumor samples from women with breast cancer confirmed that HILPDA is more abundant in tumors than in normal breast tissue, especially in triple‑negative disease, and that patients whose tumors express more HILPDA tend to fare worse. Tumors from people who did not benefit from PD‑1–blocking drugs also had more HILPDA, tying this protein directly to treatment failure.
How tumors turn the immune landscape against us
To see what HILPDA actually does inside tumors, the team engineered breast cancer cells to either overproduce or lack the protein and then grew them with human immune cells or in mice. When HILPDA was high, tumors attracted more regulatory T cells, myeloid‑derived suppressor cells, and M2‑like macrophages—immune cell types that dampen attack and help cancers hide. At the same time, the number and vigor of killer CD8 T cells and natural killer cells dropped, and their ability to release toxic molecules and inflammatory signals was blunted. Silencing HILPDA flipped this script: tumors hosted fewer suppressive cells, more active killer cells, slower growth, and fewer metastases. Crucially, when animals with low‑HILPDA tumors received anti‑PD‑1 therapy, their tumors shrank more and the mice lived longer, showing that HILPDA controls how well immunotherapy works.
Fat making, molecular armor, and immune escape
Diving deeper, the scientists uncovered how HILPDA reshapes tumor metabolism to harden immune defenses. Inside cancer cells, HILPDA latches onto a chaperone protein called HSP90 to stabilize a transcription factor, KLF5, that drives fat production. This trio boosts the synthesis of fatty acids, especially palmitate, and fills tumor cells with lipid droplets. One of these fats is then attached to PD‑L1, the checkpoint protein that sits on the tumor surface and sends a “do not attack” signal to immune cells. The team showed that adding palmitate to a specific point on PD‑L1 acts like a molecular glue: it helps PD‑L1 sit stably in the cell membrane and prevents it from being broken down. Mutating this single attachment site stripped PD‑L1 of its stability and weakened its ability to shield cancer cells, even when HILPDA was abundant. In other words, HILPDA‑driven fat production feeds a chemical tweak on PD‑L1 that turns the tumor’s immune shield from flimsy to reinforced steel.
The built‑in brake that can be pushed
Every accelerator needs a brake, and here the brake is another protein called TRIM21. The authors discovered that TRIM21 recognizes HILPDA and tags it with molecular “flags” that send it to the cell’s disposal machinery. In breast tumors from patients, TRIM21 levels tended to be low when HILPDA was high, and low TRIM21 was linked to poor outcomes, suggesting that this natural braking system is often weakened in cancer. The team then searched for drugs that could strengthen TRIM21 and identified fenretinide, a retinoid already tested in humans. In cell and animal models of triple‑negative breast cancer, fenretinide boosted TRIM21 activity, accelerated the breakdown of HILPDA, reduced fat synthesis and PD‑L1’s protective modification, and slowed tumor growth and spread. Most strikingly, combining fenretinide with anti‑PD‑1 therapy made tumors more vulnerable to immune attack, increasing killer T and natural killer cells while reducing suppressive cells in the tumor microenvironment. 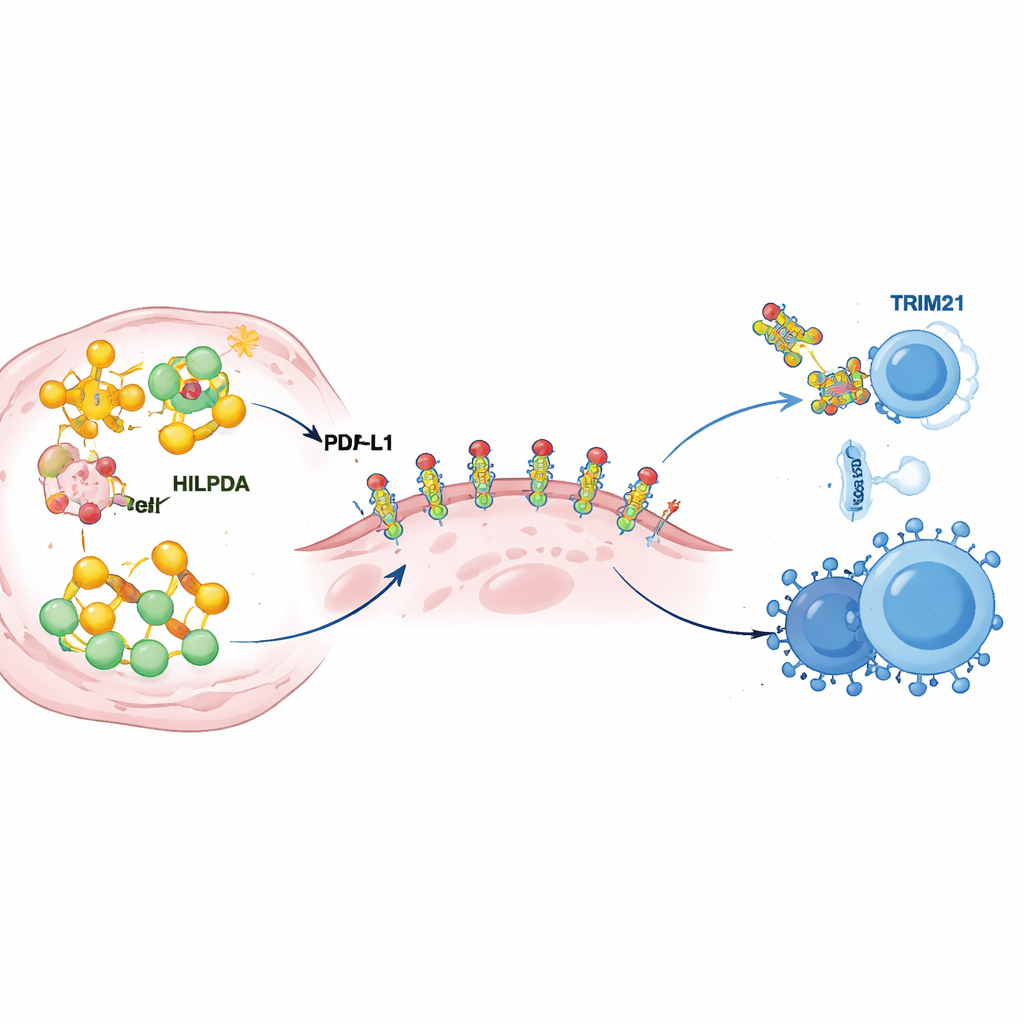
What this means for future cancer care
This work reveals a chain of events in which stressed breast cancer cells turn up HILPDA, ramp up fat production, and chemically reinforce PD‑L1 on their surface, allowing them to keep suppressing immune cells even in the face of PD‑1–blocking drugs. TRIM21 serves as an internal safeguard that can dismantle HILPDA, but it is often too weak in tumors. By finding that fenretinide can reactivate this safeguard, the study points to a practical way to combine a metabolic drug with existing immunotherapy to overcome resistance. If confirmed in clinical trials, targeting the TRIM21–HILPDA–PD‑L1 axis could give patients with hard‑to‑treat triple‑negative breast cancer a second chance at benefiting from immune‑based treatments.
Citation: Wang, X., Li, G., Wu, J. et al. TRIM21-mediated degradation of HILPDA overcomes anti-PD-1 immunotherapy resistance in breast cancer by limiting PD-L1 palmitoylation. Oncogene 45, 1338–1356 (2026). https://doi.org/10.1038/s41388-026-03728-6
Keywords: triple-negative breast cancer, immunotherapy resistance, PD-L1 palmitoylation, tumor lipid metabolism, fenretinide