Clear Sky Science · en
PMM2 interacts with TRIM28 to recruit E2F4 and promote KIFC3-mediated tumor glycolysis and colorectal cancer progression
Why This Cancer Story Matters
Colorectal cancer is one of the world’s deadliest cancers, partly because many tumors learn to hijack the body’s energy systems to fuel unchecked growth. This study uncovers how a little-known enzyme, PMM2, helps colorectal tumors burn sugar more aggressively and spread, and why that makes it a promising new target for future drugs and diagnostic tests.
A Sugar-Hungry Tumor Engine
Cancer cells often rewire the way they use glucose, favoring a high-speed, low-efficiency form of sugar burning known as glycolysis. The researchers began by comparing thousands of genes in colorectal tumor samples to nearby healthy tissue. PMM2, an enzyme usually involved in attaching sugar chains to proteins, stood out as one of the most strongly increased genes in cancer. Tumor cells with extra PMM2 grew faster, formed more colonies, and spread more readily in lab dishes, while cells in which PMM2 was switched off slowed their growth, migrated less, and were more prone to die.
How Tumor Cells Supercharge Sugar Use
When the team reduced PMM2 levels in colorectal cancer cells, the cells took up less glucose, made less ATP (their main energy currency), and released less lactate, the waste product of glycolysis. Sensitive metabolic measurements confirmed that the overall acidification of the surrounding medium dropped, while oxygen use rose, meaning the cells shifted away from turbocharged glycolysis toward more normal respiration. Key glycolysis helper proteins, PKM2 and LDHA, also fell. Surprisingly, even a catalytically “dead” version of PMM2 could still drive this sugar-hungry behavior, showing that the enzyme’s cancer role depends not on its usual chemistry, but on whom it binds to inside the cell.
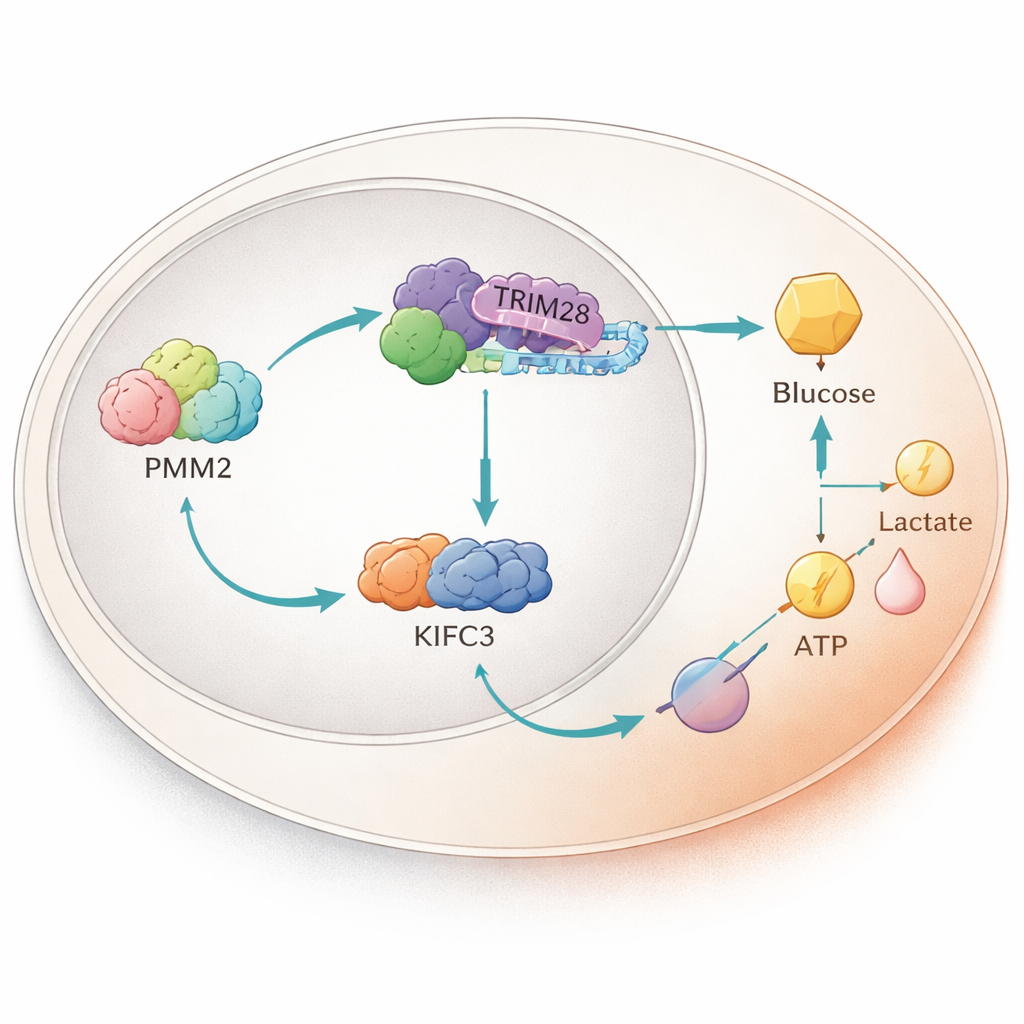
A Protein Relay Inside the Nucleus
Digging deeper, the scientists found that PMM2 physically latches onto another protein called TRIM28, which can move into the cell nucleus and influence gene activity. PMM2 helps TRIM28 accumulate in the nucleus, where TRIM28 teams up with a transcription factor, E2F4. Together, this trio boosts production of a motor protein called KIFC3 by binding to a specific stretch of its DNA control region. Experiments that deleted the PMM2 region needed for TRIM28 binding erased PMM2’s ability to ramp up glycolysis and cell growth, underscoring that this protein partnership—not PMM2’s classic enzyme function—is what powers the tumor advantage.
Turning Up a Key Metabolic Switch
KIFC3, better known for its role in moving cargo along the cell’s internal scaffolding, turned out to be a crucial metabolic switch. When the researchers lowered KIFC3 levels, colorectal cancer cells consumed less glucose, produced less ATP and lactate, and showed weaker glycolytic activity, while their oxygen use increased. Importantly, silencing KIFC3 partially canceled the glycolysis boost and growth advantage normally caused by PMM2. In mice implanted with human colorectal cancer cells, tumors with extra PMM2 grew larger, but this effect was blunted when KIFC3 was knocked down. Tumor samples from these animals showed higher levels of PMM2, KIFC3, and glycolysis markers, tying the whole chain of events together in living tissue.
From Lab Models to Patient Samples
To bring the work closer to the clinic, the team created miniature three-dimensional tumors, known as organoids, from patient colorectal cancers. Organoids with higher PMM2 and KIFC3 levels grew faster and produced more ATP and lactate than those with lower levels. Forcing organoids to make more PMM2 increased KIFC3 and glycolysis, whereas reducing PMM2 had the opposite effects. Analyses of patient tumor arrays further showed that high PMM2 levels were linked to more advanced disease, lymph node spread, and shorter overall survival, marking PMM2 as a strong candidate biomarker.
What This Means for Future Care
In simple terms, this study shows that many colorectal tumors appear to plug PMM2 into a nuclear protein relay—through TRIM28 and E2F4—to crank up KIFC3 and, in turn, their sugar-burning machinery. That metabolic surge helps cancers grow and spread. Because this route depends on protein interactions rather than PMM2’s usual enzyme job, it opens new avenues for therapy: small molecules, peptides, or degrader drugs that disrupt PMM2’s binding to TRIM28, block E2F4’s access to DNA, or dampen KIFC3 activity could all, in principle, starve tumors of their favored fuel. While such treatments are not yet available, the PMM2–TRIM28–E2F4–KIFC3 chain now stands out as a promising roadmap for more precise and metabolism-focused strategies against colorectal cancer.
Citation: Peng, Z., Ma, B., Song, Z. et al. PMM2 interacts with TRIM28 to recruit E2F4 and promote KIFC3-mediated tumor glycolysis and colorectal cancer progression. Oncogene 45, 1145–1160 (2026). https://doi.org/10.1038/s41388-026-03707-x
Keywords: colorectal cancer, tumor metabolism, glycolysis, oncogenic signaling, biomarker