Clear Sky Science · de
Interaktions‑konstruierte 3D‑Molekülgenerierung mittels Diffusionsmodell ermöglicht struktur‑basierte Pharmacophor‑Modellierung für die Wirkstoffentwicklung
Warum es so schwierig ist, bessere Medikamente zu entwerfen
Moderne Wirkstoffforschung beruht oft darauf, ein kleines Molekül dazu zu bringen, in ein Protein zu passen wie ein Schlüssel in ein Schloss. Der Schlüssel muss aber mehr können als nur passen: er muss die richtigen winzigen Anziehungskräfte ausbilden — wie schwache elektrische Wechselwirkungen und wasserabweisende Bereiche — damit das Medikament stark und selektiv gebunden bleibt. Das chemische Universum ist astronomisch groß, weit über das hinaus, was heutige Datenbanken abdecken, weshalb Forscher nach intelligenteren Wegen suchen, neue Schlüssel von Grund auf zu entwerfen und dabei diese wichtigen Kontaktmuster zu erhalten.
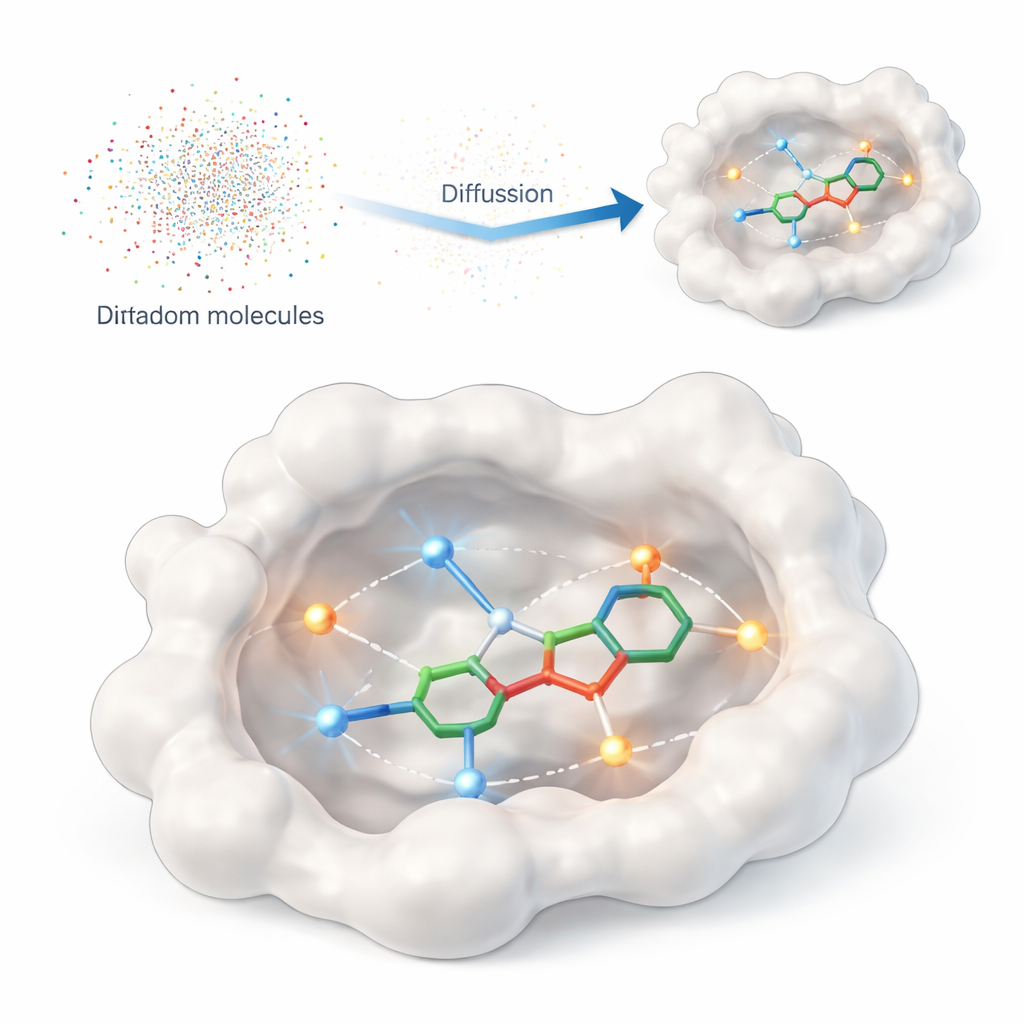
Dem Computer beibringen, was wirklich zählt
Diese Studie stellt DiffPharma vor, ein rechnerisches Framework, das dreidimensionale, wirkstoffähnliche Moleküle direkt im Bindungstaschenbereich eines Proteins erzeugt. Anstatt den Algorithmus große Kataloge vorhandener Verbindungen durchsuchen zu lassen, erschafft DiffPharma neue Moleküle Atom für Atom, geleitet von der Weise, wie sie mit dem Protein interagieren sollen. Die Methode basiert auf einer modernen Klasse generativer Modelle, den Diffusionsmodellen, die aus zufälligem Rauschen starten und dieses schrittweise „entrauschen“, bis ein strukturiertes Objekt entsteht — in diesem Fall ein 3D‑Molekül, das in der Proteintasche sitzt.
Das Händedruck‑Muster des Proteins kodieren
Um dem Modell mitzuteilen, was an der Proteinoberfläche wichtig ist, repräsentieren die Autor:innen zentrale Kontakte als kleine „Interaktionspartikel“, die entlang der Verbindungswege zwischen dem Protein und einem Referenzmolekül verteilt werden. Zwei gängige Interaktionstypen stehen dabei im Fokus: Wasserstoffbrücken, die wie richtungsorientierte Magnete zwischen bestimmten Atomen wirken, und hydrophobe Kontakte, bei denen ölige Regionen abseits des Wassers zusammenfinden. Getrennte neuronale Netze lernen Geometrie und Chemie jedes Interaktionstyps sowie die Gesamtform der Bindungsstelle; eine spezielle Fusionsarchitektur kombiniert diese Sichtweisen zu einem einheitlichen, kohärenten Bild, das die Molekülgenerierung steuert.
Wie gut ahmt es reale Bindungsmuster nach?
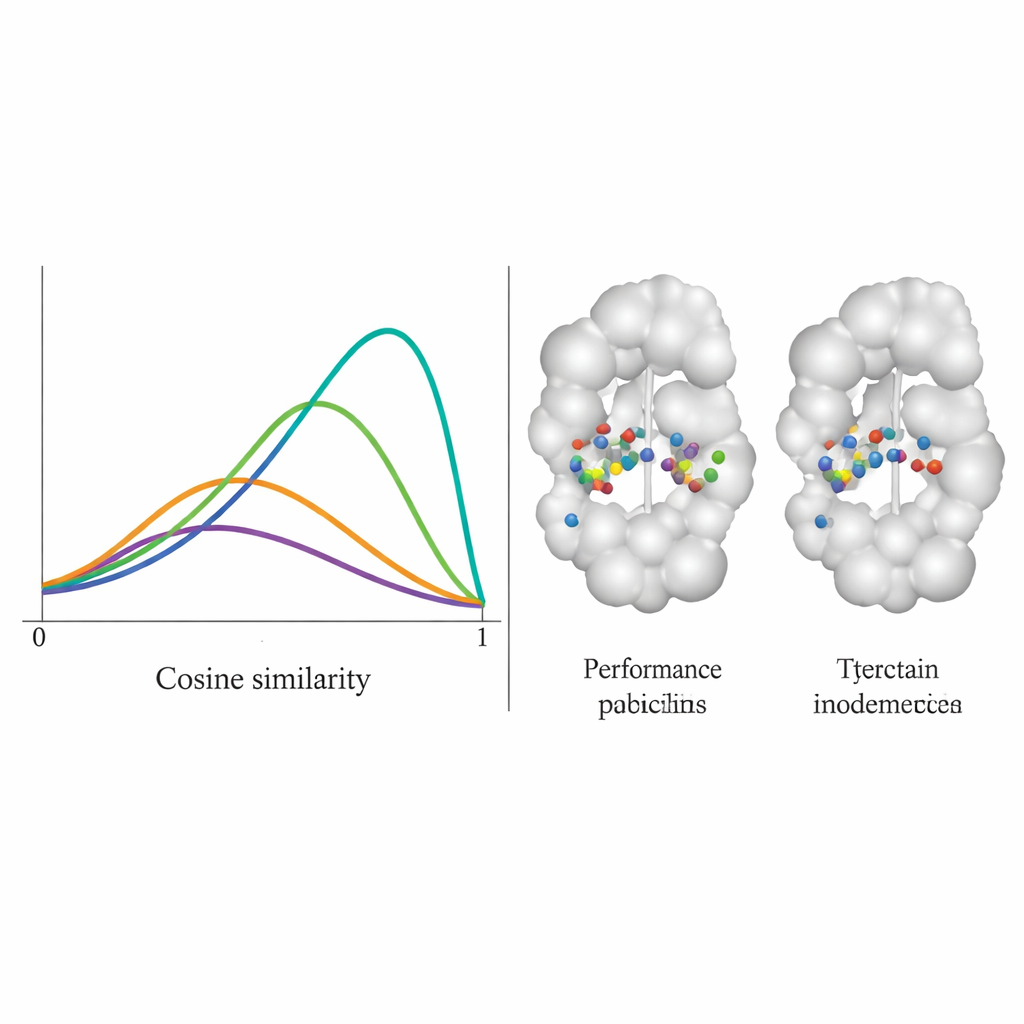
Das Team testete DiffPharma an 100 verschiedenen Protein‑Molekül‑Paaren und fragte, wie getreu neue Moleküle die ursprünglichen Kontaktmuster wiedergeben, Rest für Rest. Gemessen wurde dies mit einem Kosinus‑Ähnlichkeitswert zwischen 0 und 1, wobei 1 perfekte Übereinstimmung bedeutet. Die Verteilung von DiffPharma lag um etwa 0,9, was bedeutet, dass im Mittel dieselben Proteinreste dieselben Arten von Schlüsselkontakten ausbildeten wie in den Referenzstrukturen — deutlich besser als sechs konkurrierende Methoden. Wichtig ist, dass das Modell dies schaffte und dabei dennoch eine Vielfalt molekularer Formen erzeugte; die generierten Verbindungen wiesen realistische Bindungslängen, Winkel und eine insgesamt 3D‑Geometrie auf, wie sie für stabile reale Moleküle typisch ist.
Von der Theorie zu praktischen Wirkstoffkandidaten
Über Benchmarks hinaus prüften die Autor:innen, ob DiffPharma plausible Wirkstoffkandidaten für reale Ziele entwerfen kann. Für zwei gut untersuchte Enzyme — die AKT‑Kinase und eine β‑Lactamase, die mit Antibiotikaresistenz in Verbindung steht — erzeugte die Methode Moleküle, die die essentiellen Interaktionsmuster bekannter Liganden bewahrten, dabei aber oft unterschiedliche chemische Gerüste verwendeten, eine erwünschte Form des „Scaffold Hopping“ in der medizinischen Chemie. In einer anspruchsvolleren Fallstudie an der Hauptprotease von SARS‑CoV‑2 wurde DiffPharma mit spezifischen Interaktionsvorgaben gesteuert und anschließend mittels molekulardynamischer Simulationen und Bindungsenergie‑Schätzungen untersucht. Unter strengeren Interaktionszwängen bildeten die generierten Moleküle stabilere Komplexe und zeigten manchmal günstigere vorhergesagte Bindungsenergien als ein bekannter Referenzinhibitor. Bemerkenswerterweise entdeckte das System sogar jenes Referenzmolekül wieder — obwohl es nie im Training aufgetaucht war — allein aus der Proteinstruktur und den Interaktionsanweisungen.
Was das für zukünftige Medikamente bedeutet
Für Nicht‑Spezialist:innen lässt sich DiffPharma als intelligentes, 3D‑bewusstes Entwurfswerkzeug für Wirkstoffmoleküle verstehen: Gegeben die Form einer Proteintasche und ein gewünschtes Muster von „Händedrucken“, schlägt es chemisch sinnvolle Schlüssel vor, die passen und auf die richtige Weise interagieren. Obwohl es noch nicht jede Eigenschaft optimiert, die ein Medikament braucht — etwa Löslichkeit oder Metabolismus — bewahrt die Methode zuverlässig die entscheidende Kontaktkarte an der Proteinoberfläche und erkundet neue Bereiche des chemischen Raums jenseits heutiger Kataloge. Dieser interaktionsgeführte Ansatz kann Forschern helfen, schneller von strukturellen Daten zu krankheitsrelevanten Proteinen zu diversen, realistischen Ausgangspunkten für experimentelle Wirkstoffentwicklung zu gelangen.
Zitation: Sako, M., Yasuo, N. & Sekijima, M. Interaction-constrained 3D molecular generation using a diffusion model enables structure-based pharmacophore modeling for drug design. npj Drug Discov. 3, 8 (2026). https://doi.org/10.1038/s44386-026-00040-x
Schlüsselwörter: strukturbasierte Wirkstoffentwicklung, molekulare Generatormodelle, Pharmacophor‑Modellierung, Protein‑Ligand‑Wechselwirkungen, SARS‑CoV‑2 Hauptprotease