Clear Sky Science · de
KI-geführtes kompetitives Docking für virtuelles Screening und Vorhersage von Wirkstoffwirksamkeit
Intelligentere Suche nach neuen Arzneimitteln
Neue Wirkstoffe zu finden ist ein bisschen so, als würde man eine Nadel in einem Heuhaufen aus Millionen von Molekülen suchen. Diese Studie zeigt, wie jüngste Fortschritte in der künstlichen Intelligenz diese Suche schneller und günstiger machen können, indem sie Wissenschaftlern helfen vorherzusagen, welche Moleküle am ehesten an ein krankheitsrelevantes Protein binden und tatsächlich als Arzneimittel wirken könnten. Statt im Labor ein Chemikalie nach der anderen zu testen, setzen die Autorinnen und Autoren KI-Modelle ein, die virtuelle Wettkämpfe zwischen Molekülen austragen und die Sieger an die Spitze bringen.
Wie KI das molekulare Schloss-Schlüssel-Passen lernt
Viele moderne Medikamente wirken, indem sie in winzige Taschen auf Proteinen passen, ähnlich wie ein Schlüssel in ein Schloss. Traditionell versuchten Computerprogramme, diese Passung mit physikalischen Gleichungen zu berechnen, die die Kräfte zwischen Atomen schätzen. In den letzten Jahren haben jedoch neue KI-Systeme, sogenannte diffusion-basierte Co-Folding-Modelle – etwa AlphaFold3 und Boltz – aus riesigen Mengen bekannter Protein–Molekül-Strukturen gelernt. Diese Systeme können nun „vorstellen“, wie sich ein Protein und ein potenzielles Arzneimittel dreidimensional zusammenfalten könnten, selbst wenn keine experimentelle Struktur vorliegt. Die zentrale Frage, die die Autorinnen und Autoren untersuchen, ist, ob diese KI-Werkzeuge mehr leisten können als plausible Bilder zu erzeugen – ob sie auch gute von schlechten Wirkstoffen unterscheiden können.
Echte Binder versus Blender
Das Team testete zunächst 16 gut untersuchte Proteine sowie ein komplexeres bakterielles Enzym, die DNA-Gyrase. Für jedes Protein baten sie die KI-Modelle, sowohl bekannte aktive Inhibitoren als auch eine Reihe von nicht verwandten „Off-Target“-Molekülen in dieselbe Bindungsstelle zu platzieren. Anstatt einer einzigen Vorhersage zu vertrauen, betrachteten sie, wie konsistent die KI jedes Molekül über viele Durchläufe platzierte. Echte Inhibitoren kehrten dazu tendentiell immer wieder an denselben Ort und in dieselbe Orientierung zurück und gruppierten sich innerhalb von nur wenigen Billionstel Metern. Inaktive Moleküle wanderten weiter und lagen oft weiter vom Taschenzentrum entfernt. Diese einfache Idee – Konvergenz der Pose – erwies sich als starkes Signal dafür, dass eine Verbindung wirklich zu ihrem Proteinziel passt.
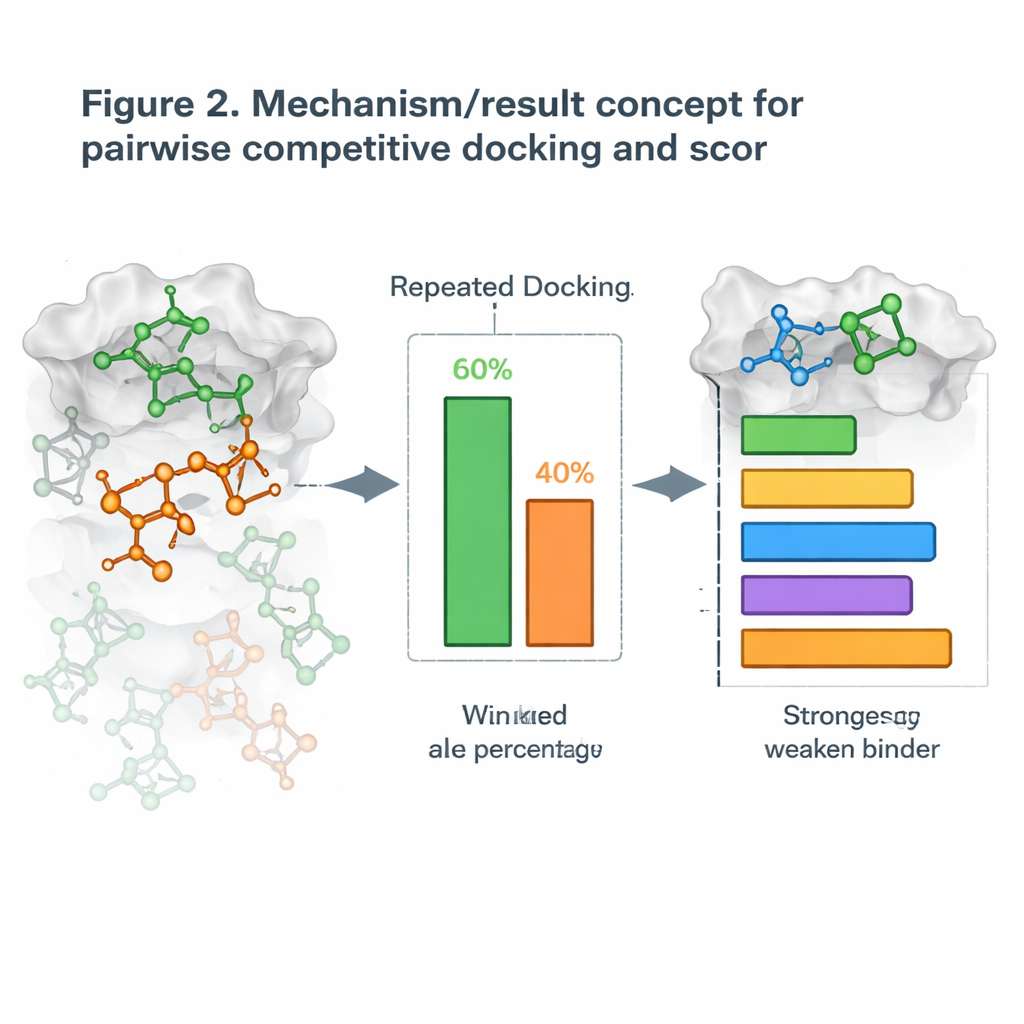
Docking als Kopf-an-Kopf-Wettkampf
Darauf aufbauend führten die Autorinnen und Autoren eine neue Strategie ein, die sie paarweises kompetitives Docking nennen. Anstatt ein Molekül nach dem anderen zu docken, docken sie zwei Kandidaten gleichzeitig zusammen mit dem Protein und lassen sie um dieselbe Tasche „konkurrieren“. Nach vielen Wiederholungen wird das Molekül, das die Stelle häufiger einnimmt, zum Gewinner dieses Duells erklärt. Durch das Ausführen aller möglichen Paarungen erstellen sie eine Gewinn-Verlust-Tabelle und berechnen für jedes Molekül einen Competitive Docking Score, ähnlich der Bewertung von Spielern in einem Round-Robin-Turnier. Wenn diese Scores mit realen Messungen der Stärke verglichen wurden, mit der die Moleküle ihre Ziele blockieren, stimmten die Ranglisten oft gut überein; bei einigen Proteinsystemen gab es nahezu perfekte Übereinstimmung.
Vom virtuellen Screening zur Entwicklung besserer Antibiotika
Die DNA-Gyrase, ein für Bakterien essentielles Enzym, diente als detaillierter Testfall. Dieses Protein besitzt mehrere Arzneimittel-Taschen, die von verschiedenen Antibiotikaklassen angegriffen werden, einschließlich weit verbreiteter Fluorchinolone. Die KI-Modelle konnten in der Regel jede Wirkstoffklasse in die korrekte Tasche platzieren, und die Competitive Docking Scores folgten ungefähr ihren gemessenen Potenzen. Die Autorinnen und Autoren skalierten die Methode dann auf ein virtuelles Screening von mehr als 3.000 zugelassenen Arzneimitteln, um zu ermitteln, welche Moleküle am besten um die Fluorchinolon-Stelle konkurrierten. Ihre zweistufige Strategie – zuerst ein „All-at-once“-Wettbewerb, um wahrscheinliche Gewinner auszuwählen, dann Filterung nach der Dichte ihrer Clusterung in der Tasche – reicherte echte Fluorchinolone stark an und sortierte schwächere Kandidaten aus. Schließlich nutzten sie einen KI-getriebenen Molekülgenerator, um neue fluorchinolonähnliche Strukturen vorzuschlagen, und wandten das competitive Docking an, um eine Handvoll mit noch besser vorhergesagter Bindung und akzeptablen arzneimittelähnlichen Eigenschaften zu finden.
Versprechen, Grenzen und Bedeutung für Patientinnen und Patienten
Die Studie zeigt, dass moderne KI-Modelle mehr können als plausible Protein–Wirkstoff-Strukturen zu zeichnen: Wenn sie in einem kompetitiven Rahmen betrieben werden, können sie helfen, Verbindungen so zu bewerten, dass die Reihenfolge häufig mit realen experimentellen Daten übereinstimmt. Das ersetzt nicht die Laborarbeit – die Leistung hängt weiterhin stark vom jeweiligen Protein ab, einige Taschen werden falsch vorhergesagt, und KI-Modelle können bei sehr großen oder ungewöhnlichen Molekülen versagen. Aber mit der Verbesserung dieser Modelle und ihrer Trainingsdaten könnten Ansätze wie das paarweise kompetitive Docking die frühe Wirkstoffforschung deutlich effizienter machen. Für Patientinnen und Patienten könnte das langfristig schnellere Entwicklung gezielter Medikamente bedeuten, einschließlich neuer Antibiotika, die mit resistenten Bakterien Schritt halten.
Zitation: Mirgaux, M., Barcelli, V., Chua, A.C.Y. et al. AI-guided competitive docking for virtual screening and compound efficacy prediction. npj Drug Discov. 3, 6 (2026). https://doi.org/10.1038/s44386-026-00039-4
Schlüsselwörter: KI Wirkstoffforschung, virtuelles Screening, molekulares Docking, Protein-Ligand-Bindung, Antibiotikadesign