Clear Sky Science · de
Nichtgleichgewichts-Chemomechanische Umwandlung von ATP‑getriebener Proteinentfaltung im 26S‑Proteasom
Wie eine winzige Maschine unsere Proteine in Schach hält
In jeder Zelle zerstört ein mikroskopischer Schredder, das 26S‑Proteasom, fortlaufend beschädigte oder nicht mehr benötigte Proteine und erhält so die Zellgesundheit. Im Zentrum dieser Maschine sitzt ein ringförmiger Motor, der chemischen Treibstoff (ATP) verbraucht, um Proteine zu erfassen, zu entfalten und in eine zentrale Kammer zu ziehen, wo sie zerkleinert werden. Diese Arbeit nutzt fortgeschrittene Computersimulationen, um zu zeigen, wie der Motor chemische Energie in mechanische Bewegung umwandelt, und liefert ein detailliertes, quantitatives Bild eines Prozesses, der Altern, neurologische Erkrankungen, Immunität und Krebs zugrunde liegt.
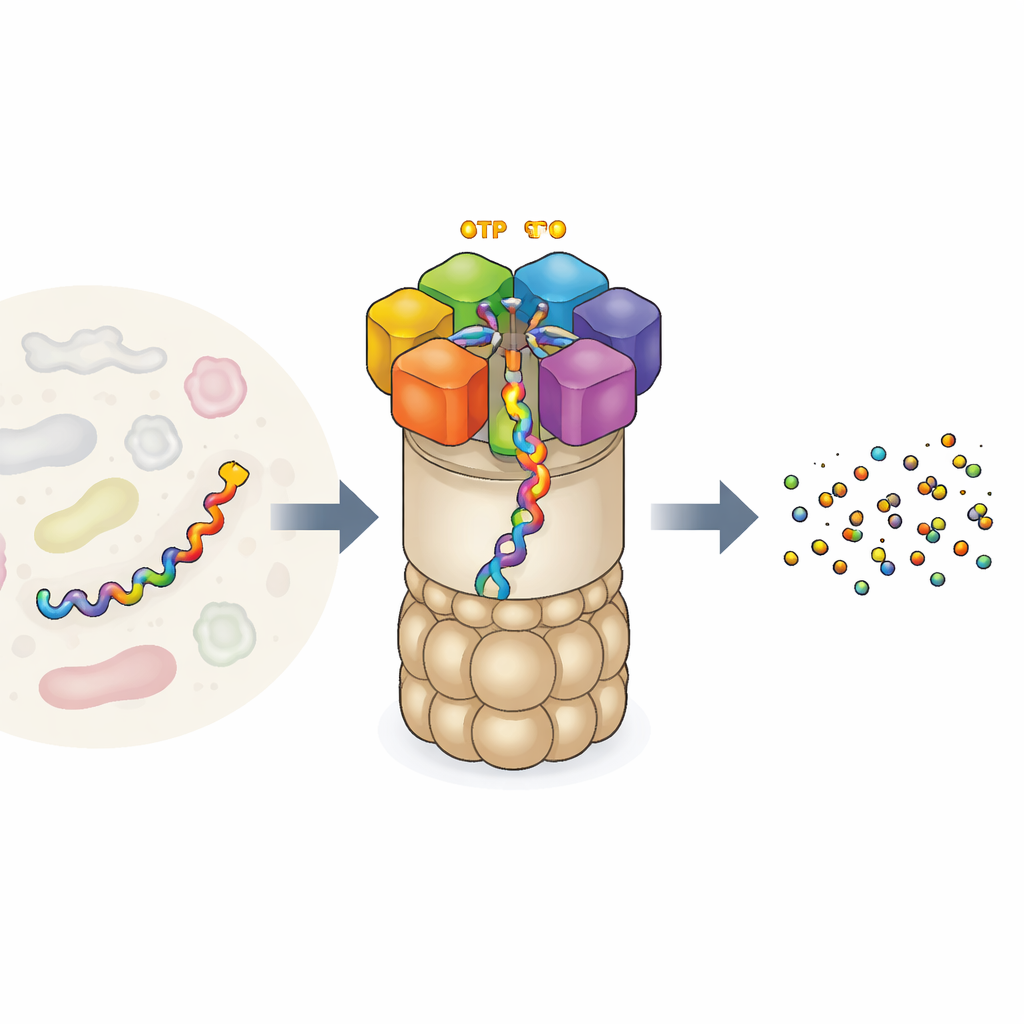
Die Recyclingfabrik der Zelle für Proteine
Das 26S‑Proteasom ist eine der größten und komplexesten Proteinkomplexe in unseren Zellen. Es besteht aus einem faßförmigen Kern, der Proteine zerschneidet, und einer regulatorischen Kappe, die erkennt, welche Proteine abgebaut werden sollen. Am Eingang sitzt ein Ring aus sechs unterschiedlichen Motoruntereinheiten. Jede Einheit kann ATP binden — die universelle Energiewährung der Zelle — und nutzt diese Energie, um an einer Proteinkette zu greifen und sie in den Kern zu ziehen. Dieser Motor arbeitet nicht zufällig: Frühere Experimente deuteten darauf hin, dass die sechs Untereinheiten koordiniert in einer „Hand‑über‑Hand“-Manier zusammenwirken und das Protein wie Kletterer an einem Seil weiterreichen. Experimente lieferten jedoch nur einige wenige Momentaufnahmen dieses Ablaufs, sodass Fragen nach der gesamten Bewegungssequenz und danach, wie genau das Treibstoffverbrennen mit mechanischer Arbeit verknüpft ist, offen blieben.
Simulation eines molekularen Tauziehens
Die Autoren entwickelten ein probabilistisches Computermodell, das den Motor als System behandelt, das zwischen vielen möglichen Konformationen hin‑ und herspringt, während ATP‑Moleküle und deren Produkte binden und wieder abgehen. Sie definierten 30 Hauptkonformationen des Rings, in denen eine, zwei oder drei Motoruntereinheiten ihren Griff am Protein verlieren, sowie einen speziellen, fest geschlossenen Zustand, in dem alle sechs halten. Mit einem standardisierten Algorithmus zur Simulation zufälliger chemischer Ereignisse verfolgten sie Hunderttausende von Schritten, in denen ATP‑Bindung, ATP‑Zerlegung und Formänderungen des Motors stattfinden. Aus diesen Läufen konnten sie vorhersagen, wie schnell ein Protein unter verschiedenen Bedingungen durchgezogen wird, etwa bei variierenden Mengen an ATP, seinem verbrauchten Produkt ADP und einem in Experimenten häufig verwendeten nicht‑spaltbaren ATP‑Mimetikum.
Wenn zu viel Treibstoff den Motor bremst
Die Simulationen rekonstruierten mehrere rätselhafte experimentelle Befunde. Steigt die ATP‑Konzentration von niedrigen Werten an, zieht der Motor Proteine schneller, weil das Treibstoffbinden der langsamste Schritt ist. Jenseits von etwa 1 Millimolar ATP erreicht die Geschwindigkeit jedoch ein Maximum und fällt dann wieder ab: Der Ring verbringt mehr Zeit in einer verstopften, nicht‑translozierenden Konformation, in der alle sechs Untereinheiten mit ATP besetzt sind, das Protein sich aber nicht bewegt. Zugabe von ADP oder des nicht‑spaltbaren ATP‑Analogons verlangsamt den Motor kontinuierlich, weil diese Moleküle mit ATP um Bindungsstellen konkurrieren, aber den vollen Kraftstoß nicht abschließen können. Das Modell sagt auch voraus, wie sich der Motor verhält, wenn er auf sehr dicht gefaltete Bereiche eines Proteins trifft, die als Hindernisse wirken. In solchen Fällen kämpft der Motor länger gegen den Widerstand, und die Gesamtabbaugeschwindigkeit sinkt — im Einklang mit Messungen an künstlich stabilisierten Proteindomänen.
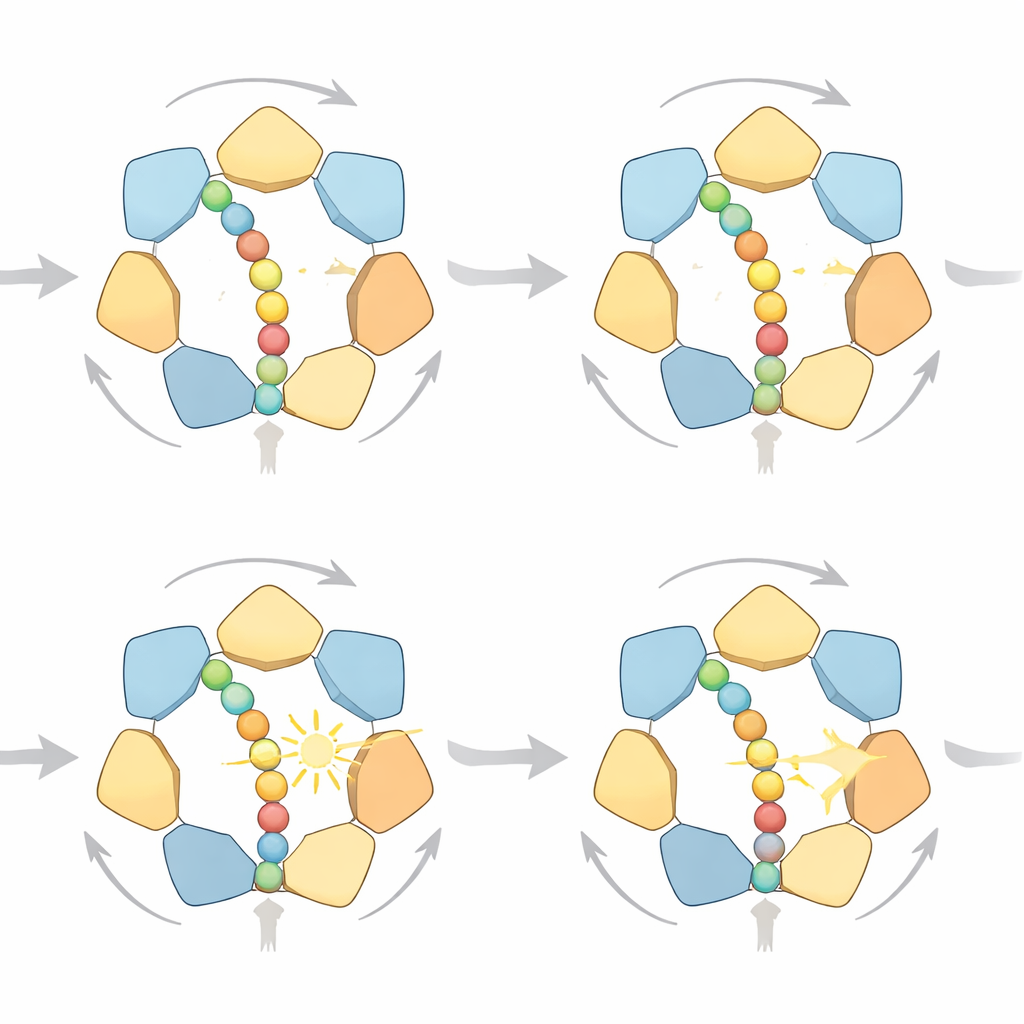
Viele Arten, einen Schritt zu machen
Bei detaillierter Untersuchung der simulierten Trajektorien fanden die Forscher heraus, dass der Ring keinem einzigen starren Zyklus folgt. Stattdessen existieren mehrere wahrscheinliche Pfade, die alle derselben richtungsgebenden "Hand‑über‑Hand"‑Regel gehorchen: Untereinheiten nahe dem Ausgang des Rings lassen nach ATP‑Hydrolyse das Protein los, bewegen sich an die Spitze einer spiralförmigen Treppe von Untereinheiten und greifen die Kette weiter oben wieder. Manchmal macht das Protein einen kleinen Einzelschritt, manchmal zwei, abhängig davon, wie viele Untereinheiten gleichzeitig loslassen. Bei reichlich Treibstoff dominieren Einzelschritte, weil sie weniger Arbeit gegen entgegengesetzte Kräfte verschwenden; bei knapper werdendem Treibstoff sagt das Modell häufigere Zwei‑Schritt‑Sprünge voraus. Die Simulationen verbinden außerdem mechanische Last mit dem chemischen Zustand des Motors: Wenn der Widerstand zunimmt und das Protein ins Stocken gerät, häuft sich tendenziell ADP in mehr der sechs Bindungsstellen an — genau das zeigen hochaufgelöste Strukturstudien.
Energieverbrauch und gemeinsame Gestaltungsprinzipien
Das Modell erlaubt es den Autoren, abzubilden, wie die Energie des Motors beim ATP‑Abbau ansteigt und wieder abfällt, wenn diese Energie in Bewegung umgesetzt wird. Sie berechnen eine Wirkungsgradkurve, die zeigt, dass der Motor bei einer mittleren Gegenkraft am besten arbeitet: Zu wenig Widerstand führt zu verschwenderischem ATP‑Verbrauch, zu viel bringt den Motor fast zum Stillstand. Verglichen mit Daten verwandter Proteinabbau‑Maschinen aus Bakterien und Hefen zeigten sich sehr ähnliche Trends, wie ein nicht‑spaltbares ATP‑Mimetikum diese Motoren verlangsamt. Das legt nahe, dass viele Mitglieder dieser Familie ringförmiger Enzyme wahrscheinlich einen gemeinsamen, konservierten Mechanismus zum Ziehen an Proteinen teilen.
Warum das für Gesundheit und Krankheit wichtig ist
Indem verstreute strukturelle Momentaufnahmen und biochemische Messungen in einen einzigen, prüfbaren Rahmen überführt werden, zeigt diese Arbeit quantitativ, wie ein winziger molekularer Motor chemischen Treibstoff in Kraft umsetzt, um die Proteine der Zelle zu recyceln. Das Modell erklärt nicht nur eine Vielzahl vorhandener Experimente, sondern macht auch Vorhersagen darüber, wie Änderungen der Treibstoffniveaus, der mechanischen Last oder Mutationen im Motor den Proteinabbau verändern könnten. Da ähnliche Maschinen in allen Lebensformen wirken und eine zentrale Rolle bei Erkrankungen von Neurodegeneration bis Krebs spielen, könnte das Verständnis ihres Innenlebens auf dieser Ebene letztlich die Entwicklung von Wirkstoffen leiten, die diese mikroskopischen Schredder gezielt drosseln, verstärken oder selektiv blockieren.
Zitation: Wu, D., Ouyang, Q., Wang, H. et al. Nonequilibrium chemomechanical transduction of ATP-driven protein unfolding in the 26S proteasome. npj Biol. Phys. Mech. 3, 4 (2026). https://doi.org/10.1038/s44341-026-00034-w
Schlüsselwörter: Proteasom, AAA+ ATPase‑Motor, Proteinabbau, molekulare Maschinen, Chemomechanische Kopplung