Clear Sky Science · de
Energieeffizientes wissenschaftliches Rechnen mit chemischen Reservoiren
Warum es wichtig ist, Chemie in Rechnen zu verwandeln
Moderne Supercomputer verbrauchen enorme Mengen Strom, um Klima zu simulieren, neue Medikamente zu entwerfen oder künstliche Intelligenz zu trainieren. Da wir die physikalischen Grenzen herkömmlicher Chips erreichen, wird es immer schwieriger und teurer, mehr Leistung pro Watt herauszuholen. Dieses Paper untersucht einen radikal anderen Weg: reale chemische Reaktionen als Motor wissenschaftlicher Rechnungen zu nutzen. Indem Moleküle und ihre Wechselwirkungen als die beweglichen Teile eines Rechners betrachtet werden, skizzieren die Autorinnen und Autoren, wie künftige Maschinen komplexe Gleichungen mit deutlich geringerem Energieaufwand als heutige digitale Hardware lösen könnten.
Von lebenden Zellen zu chemischen Rechnern
Lebende Zellen sind Meister der Problemlösung. Sie koordinieren ständig tausende Reaktionen, um sich anzupassen, zu wachsen und zu überleben — und das bei bemerkenswert geringem Energieeinsatz. Im Zentrum dieses Verhaltens stehen chemische Reaktionsnetze: miteinander verbundene Reaktionen, deren Raten und Konzentrationen sich über die Zeit ändern. Diese Netze lassen sich durch gewöhnliche Differentialgleichungen beschreiben, dieselbe mathematische Sprache, mit der alles modelliert wird, von Epidemien bis zu turbulenten Strömungen. Die Erkenntnis hinter dieser Arbeit ist, dass, wenn die Chemie diese Gleichungen ohnehin befolgt, man sie direkt dazu nutzen könnte, die Berechnungen durchzuführen, die Forscher heute auf Siliziumchips ausführen.
Wie aus Gleichungen Reaktionsnetze werden
Die Autorinnen und Autoren stellen ChemComp vor, ein Software-Framework, das ein System von Differentialgleichungen nimmt und systematisch in ein abstraktes Netzwerk von Reaktionen überführt. ChemComp verwendet moderne Compiler-Technologie, um ein mathematisches Problem in Muster zu zerlegen, die durch idealisierte Reaktionen darstellbar sind, und organisiert diese dann in ein Netz mit klar definierten Spezies, Verbindungen und Raten. Diese abstrakten Reaktionen entsprechen noch keinen realen Molekülen, bilden aber einen Bauplan für einen chemischen Rechner. Das Framework kann anschließend in biochemischen Reaktionsdatenbanken nach realen Reaktionsmotiven suchen, die sich ähnlich verhalten, wobei Optionen bevorzugt werden, die im Labor praktisch, sicher und potenziell energieeffizient sind.
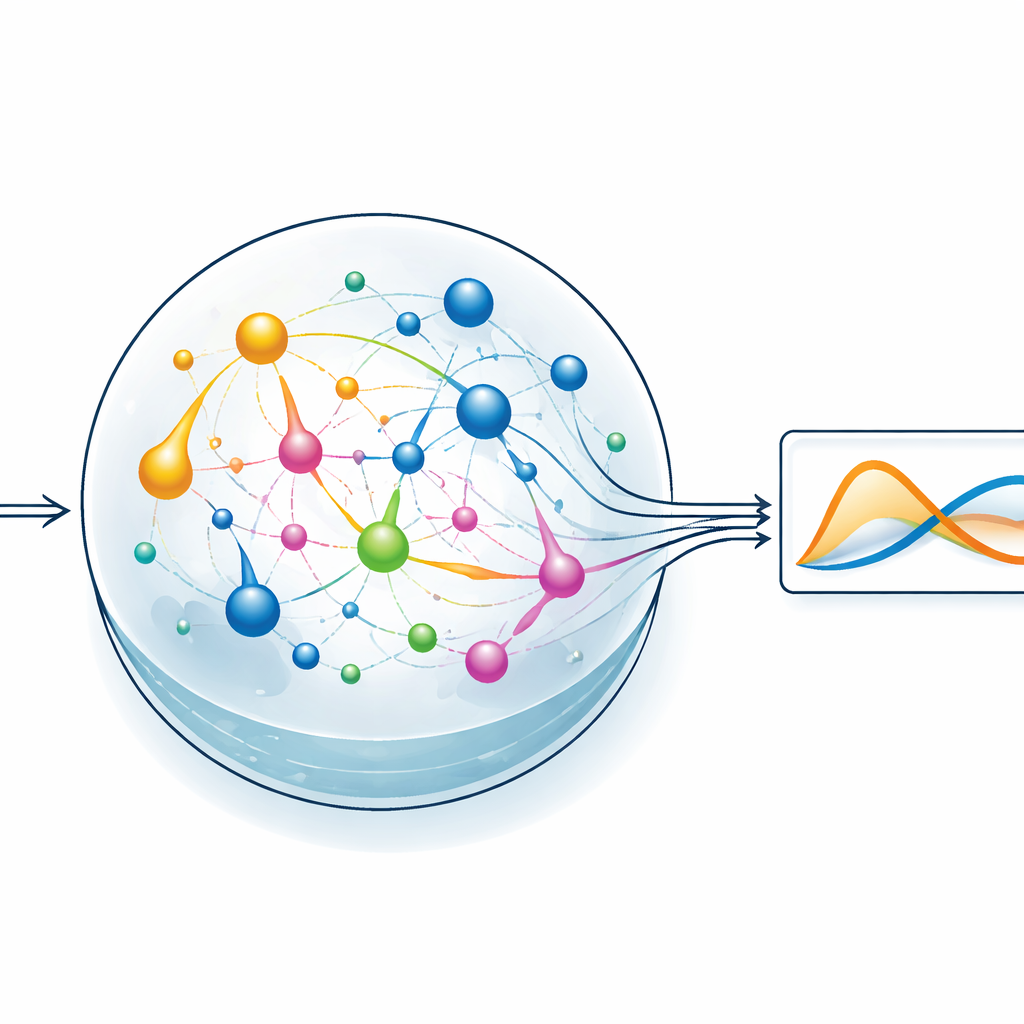
Das schwere Rechnen dem chemischen Reservoir überlassen
Um die Idee zu testen, konzentriert sich das Team auf einen Stil des maschinellen Lernens namens Reservoir-Computing. Dabei verwandelt ein festes, dynamisches System ein Eingangssignal in ein reiches, verästeltes Muster interner Aktivität, und nur eine einfache Ausleseschicht wird trainiert, um die gewünschte Ausgabe zu erzeugen. In ChemComps Variante ist das Reservoir eine Menge von Reaktionen in einem gut durchmischten Gefäß; die sich ändernden Konzentrationen der Chemikalien bilden die internen Zustände. Die Autorinnen und Autoren kompilieren ein klassisches Zwei-Variablen-System, bekannt als Sel’kov–Schnakenberg-Modell — ursprünglich zur Untersuchung von Oszillationen im Stoffwechsel — in Kandidaten-Reaktionsnetze. Sie simulieren dann, wie diese Netze im Zeitverlauf auf Zu- und Abflüsse von Chemikalien im Gefäß reagieren, und verwenden einfache lineare Regression, um die Konzentrationsverläufe zu einer Annäherung an die Zielösung zu kombinieren.
Tests einfacher und reichhaltiger chemischer Netze
Die Forschenden vergleichen zwei Kandidaten für das Reservoir: eines mit nur zwei chemischen Spezies und zwei Reaktionen und ein anderes mit fünf Spezies und fünf Reaktionen. Beide Netze erhalten geeignete Anfangskonzentrationen und Flussraten und werden dann im Betrieb simuliert. Schon das kleinere System kann das oszillierende Verhalten der Zielgleichungen grob nachbilden, aber das größere Netzwerk schneidet spürbar besser ab und reduziert den Fehler sowohl beim Training als auch beim Testen. Durch das Durchscannen verschiedener Anfangskonzentrationen und Reaktionsraten kartieren die Autorinnen und Autoren Bereiche, in denen das chemische System die gewünschte Dynamik am engsten nachbildet. Jede Reaktion wirkt dabei effektiv wie eine Basisfunktion in einem Kurvenanpassungsproblem: Je vielfältiger die verfügbaren Reaktionen, desto leichter lässt sich komplexes Verhalten annähern — allerdings zu Lasten einer höheren Systemkomplexität.
Weg zu energiearmen Rechnersystemen im Labor
Über Simulationen hinaus blickt das Paper auf praktische Geräte. Es diskutiert, wie die Wahl der Reaktionen Energieverbrauch, Steuerbarkeit mit Enzymen oder Katalysatoren und die Möglichkeit, wichtige Spezies in Echtzeit zu messen — etwa mit optischen oder elektrochemischen Methoden — in Einklang bringen muss. Die Autorinnen und Autoren schlagen vor, dass künftige mikrofluidische Plattformen gezielt ausgewählte Reaktionsnetze beherbergen könnten, mit räumlicher Kontrolle der Eingänge und integrierter Sensorik. Obwohl viele ingenieurtechnische Herausforderungen bleiben — von der Abbildung von Gleichungen auf reale Chemie bis zum Umgang mit Rauschen und Messgrenzen — zeigt die Studie, dass bereits bescheidene Reaktionssysteme die Lösungen gekoppelte Differentialgleichungen emulieren können. Für die interessierte Leserschaft lautet die Kernbotschaft: Chemie selbst kann als analoger Rechner dienen und eröffnet damit einen Weg zu wissenschaftlichen Berechnungen, die auf den energieeffizienten Prozessen beruhen, die die Natur über Milliarden von Jahren perfektioniert hat.
Zitation: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Schlüsselwörter: chemische Datenverarbeitung, energieeffizientes Rechnen, Reservoir-Computing, chemische Reaktionsnetze, gewöhnliche Differentialgleichungen