Clear Sky Science · de
Ein End-to-End-Rahmenwerk für Reaktivität in heterogener Katalyse
Warum schnellere Katalysatorentwicklung wichtig ist
Die moderne Gesellschaft ist auf Katalysatoren angewiesen, um Treibstoffe, Kunststoffe, Düngemittel und zahllose Alltagsprodukte herzustellen. Bessere Katalysatoren zu finden, gleicht jedoch oft der Suche nach der Nadel im Heuhaufen, weil jedes Material Hunderte bis Tausende möglicher mikroskopischer Reaktionen gleichzeitig fördern kann. Dieser Artikel stellt CARE vor, ein neues rechnerisches Rahmenwerk, das mithilfe intelligenter Regeln und maschinellen Lernens diese verwobenen Reaktionsnetze deutlich schneller und vollständiger abbildet und simuliert als bisher. Dadurch kann es sauberere Energietechnologien und effizientere chemische Prozesse voranbringen und zugleich die Rechenkosten drastisch senken.
Verzettelte Reaktionspfade entwirren
Auf der Oberfläche eines festen Katalysators folgen einlaufende Moleküle nicht einfach einem einzigen geradlinigen Pfad vom Edukt zum Produkt. Stattdessen durchlaufen sie ein Labyrinth kurzlebiger Zwischenstufen und konkurrierender Wege. Traditionelle Computerverfahren verlassen sich auf menschliche Intuition, um eine begrenzte Auswahl möglicher Schritte zu treffen und diese dann mit Quantenberechnungen energetisch zu bewerten. Das funktioniert bei kleinen Netzwerken, bricht aber schnell zusammen, sobald die Systeme komplexer werden, und übersieht seltene Wege, die langfristig Aktivität, Deaktivierung oder Selektivität bestimmen können. CARE begegnet dieser Herausforderung, indem es automatisch sehr große Reaktionsnetzwerke aus einfachen Bauregeln konstruiert und sicherstellt, dass alle plausiblen Bindungsauf- und -abbauereignisse zwischen Kohlenstoff, Wasserstoff und Sauerstoff eingeschlossen werden – auch solche, die Chemiker*innen sonst möglicherweise verwerfen würden.
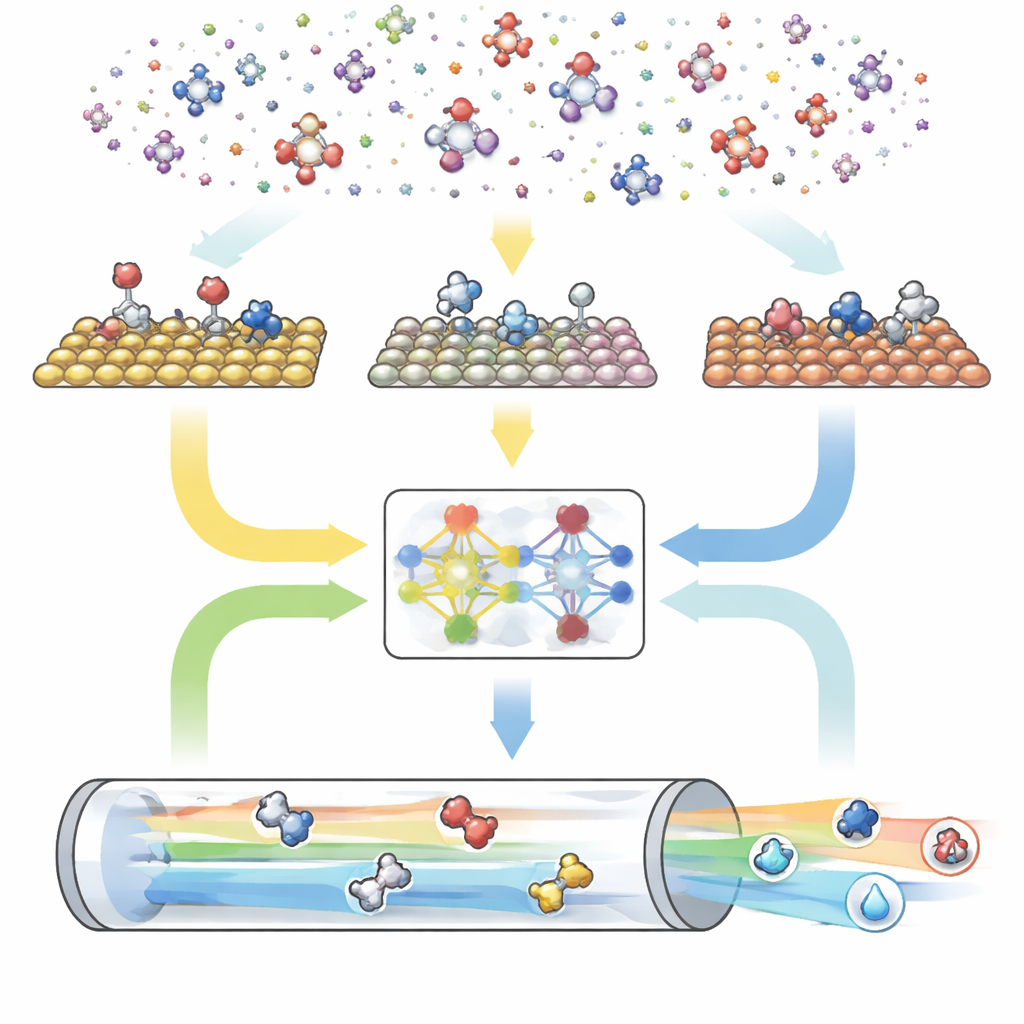
Eine dreiteilige digitale Maschine für Reaktionen
CARE ist als End-to-End-Pipeline mit drei Hauptmodulen aufgebaut. Zuerst definiert ein regelbasierter Generator den „chemischen Raum“, indem er die maximale Zahl von Kohlenstoff- und Sauerstoffatomen festlegt und dann einfache Vorlagen anwendet, um alle passenden Moleküle und ihre oberflächengebundenen Formen zu erzeugen. Das zweite Modul zur Energieabschätzung ruft moderne Modelle des maschinellen Lernens auf – insbesondere ein graphneuronales Netzwerk namens GAME-Net-UQ –, um die Energien von Zwischenstufen und Übergangszuständen auf vielen Metalloberflächen zu schätzen. Dieses Modell behandelt jede Struktur als Netzwerk aus Atomen und Bindungen, liefert sowohl Energie als auch Unsicherheit und erreicht eine Genauigkeit im Bereich weniger Zehntel Elektronenvolt bei zugleich geringerem Rechenaufwand und hoher Geschwindigkeit. Drittens nutzt ein mikrokinetischer Löser diese Energien, um zu berechnen, wie alle Reaktionen gemeinsam unter realistischen Bedingungen von Temperatur, Druck, Spannung und pH ablaufen, und sagt Gesamtreaktionsraten, Oberflächenbedeckungen und Produktselektivität voraus.
Praktische Tests: Brennstoffe und Klimachemie
Um zu zeigen, dass CARE keine bloße theoretische Übung ist, wenden die Autor*innen es auf drei industriell relevante Probleme mit steigender Schwierigkeit an. Für die Methanoldesorption – eine Reaktion, die für die Wasserstoffspeicherung wichtig ist – erzeugen sie ein überschaubares Netzwerk und bewerten es über viele Metallkatalysatoren und Kristallflächen hinweg. CARE reproduziert den bekannten „Vulkan“-Trend in der Aktivität und identifiziert korrekt Ruthenium als einen der besten Kandidaten, im Einklang mit Experimenten, jedoch mit einem Bruchteil der Rechenzeit, die vollständige Quantenberechnungen erfordern. Anschließend wenden sie sich der elektrochemischen Umsetzung von Kohlendioxid auf Kupfer zu und untersuchen, wie Drei-Kohlenstoff-Produkte wie 1-Propanol und Propen entstehen. Indem spezielle Schritte berücksichtigt werden, die Protonen, Elektronen und Lösungseinflüsse einbeziehen, erfasst CARE, wie pH-Wert und angelegte Spannung Pfade verschieben, und sagt korrekt voraus, dass 1-Propanol gegenüber Propen begünstigt ist – ein Ergebnis, das detaillierte frühere Studien widerspiegelt.
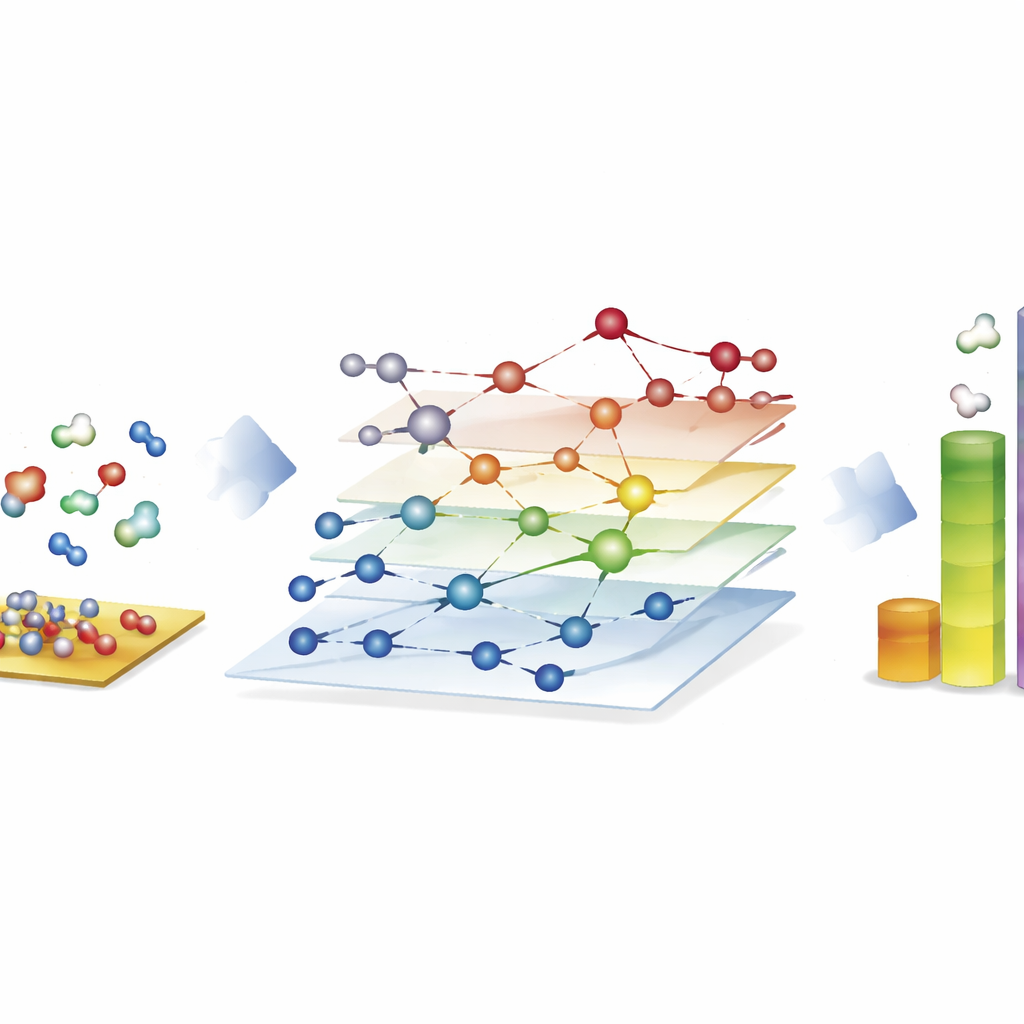
Große Reaktionsnetze für synthetische Brennstoffe erkunden
Die eindrücklichste Demonstration liefert der Fischer–Tropsch-Prozess, der Kohlenmonoxid und Wasserstoff in langkettige Kohlenwasserstoffe für Treibstoffe und Chemikalien umwandelt. Hier konstruieren die Autor*innen Netzwerke mit nahezu 40.000 Oberflächenspezies und etwa 370.000 elementaren Reaktionen – weit über das hinaus, was traditionelle quantenbasierte Studien vollständig untersuchen können. Mit CARE bewerten sie alle Zwischenstufen und wichtigen Reaktionsbarrieren auf Kobalt-, Eisen-, Nickel- und Rutheniumoberflächen in nur wenigen Stunden auf Standardhardware, was einer Beschleunigung um etwa den Faktor eine Million gegenüber direkten Quantenberechnungen entspricht. Mikrokinetische Simulationen dieser Netzwerke reproduzieren bekannte Trends: Kobalt und Eisen bilden bevorzugt längere Kohlenwasserstoffketten, Eisen erzeugt durch Nebenreaktionen mehr Kohlendioxid, und Nickel tendiert zu stärkerer Hydrierung. Obwohl einige Details, etwa Methanausbeuten, noch unvollständig sind, zeigt das Rahmenwerk, welche bindungsbildenden Schritte das Kettenwachstum dominieren und wo Modelle noch verbessert werden müssen.
Was das für zukünftige Katalysatoren bedeutet
Für Nichtfachleute ist die zentrale Botschaft, dass CARE eine praktikable Methode bietet, enorme Reaktionsräume auf katalytischen Oberflächen zu erkunden, die zuvor unerreichbar waren. Durch die Automatisierung der Netzwerkgenerierung, das Einsetzen schneller maschineller Lern-„Surrogat“-Modelle für Quantenenergien und das effiziente Lösen der resultierenden Kinetik kann es Kandidatenkatalysatoren ranken, vielversprechende Betriebsbedingungen identifizieren und unerwartete Pfade mit deutlich weniger menschlicher Voreingenommenheit und Rechenaufwand aufdecken. Zwar nennen die Autor*innen noch verbleibende Herausforderungen – etwa den besseren Umgang mit überfüllten Oberflächen, Lösungsmittelwirkungen und noch größeren Netzwerken –, doch die Arbeit weist in eine Zukunft, in der Computer komplexe Reaktionen schnell screenen können, von CO2-Reduktion über Kunststoffrecycling bis hin zur Aufwertung von Biomasse, und damit Experimente auf die vielversprechendsten Ideen lenken, statt Entdeckung dem Versuch-und-Irrtum zu überlassen.
Zitation: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Schlüsselwörter: heterogene Katalyse, Reaktionsnetzwerke, maschinelles Lernen, Mikrokinetische Modellierung, Fischer–Tropsch-Synthese