Clear Sky Science · de
Hämatopoetische Expression von cIAP2 treibt Entzündung und Herzversagen nach Myokardinfarkt voran
Warum das Beruhigen der Entzündung nach einem Herzinfarkt wichtig ist
Ein Herzinfarkt zu überleben ist nur der Anfang. In den Tagen und Wochen danach eilt das Immunsystem herbei, um geschädigtes Gewebe zu beseitigen und die Reparatur einzuleiten. Wenn diese Entzündungsreaktion jedoch zu heftig wird oder zu lange anhält, kann aus hilfreicher Heilung bleibender Herzschaden und Herzversagen entstehen. Diese Studie enthüllt einen wichtigen molekularen Schalter in blutbildenden Immunzellen, der dieses entzündliche Feuer am Lodern hält — und zeigt, dass das Ausschalten dieses Schalters das Herz in experimentellen Modellen schützen kann.
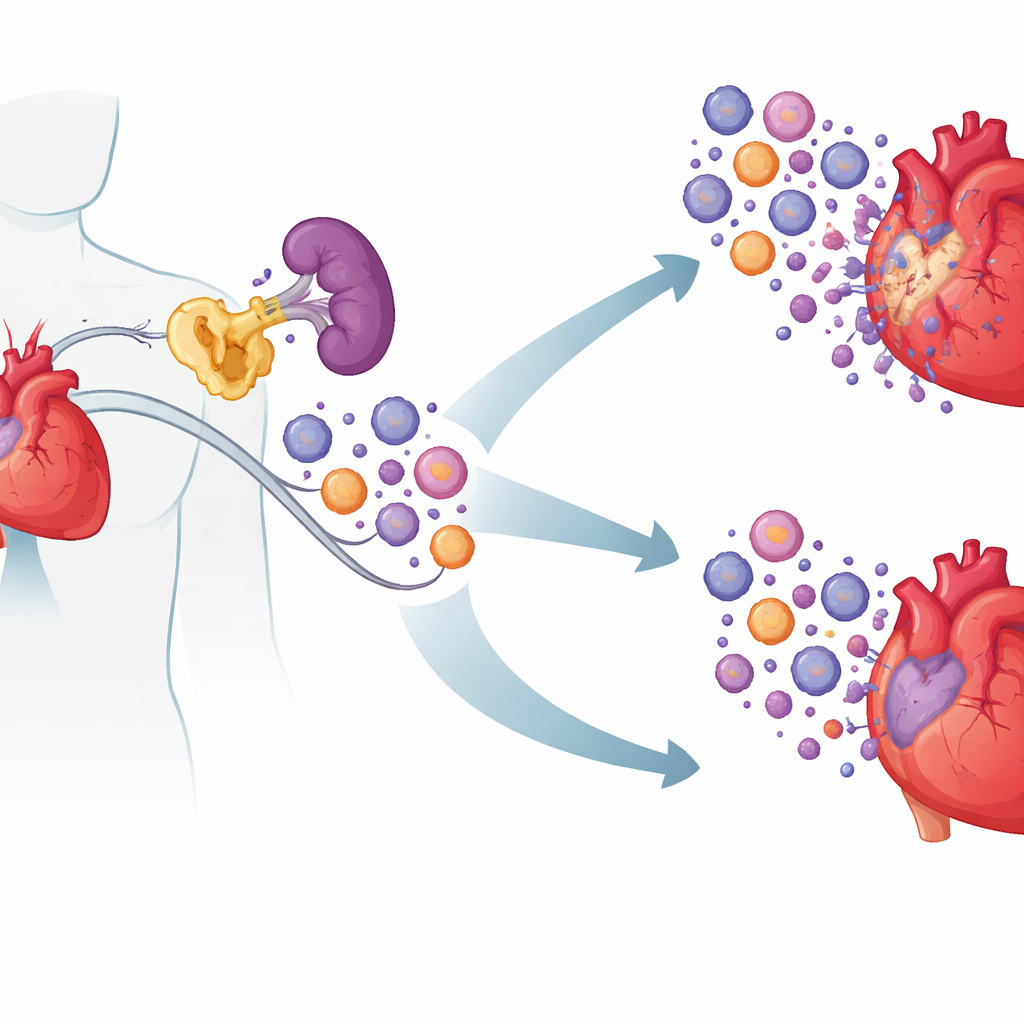
Ein verborgener Übeltäter in Immunzellen
Die Forscher konzentrierten sich auf ein Protein namens cIAP2, das vor allem dafür bekannt ist, Krebszellen das Überleben zu ermöglichen. Anhand von Blutproben von Patientinnen und Patienten mit akuten Herzerkrankungen stellten sie fest, dass die cIAP2-Spiegel bei Personen mit kürzlich zurückliegenden Herzinfarkten und ischämischem Herzversagen höher waren als bei gesunden Probanden oder Patienten mit stabiler koronarer Erkrankung. Herzgewebe von Menschen und Mäusen zeigte dasselbe Muster: cIAP2 stieg kurz nach einem Herzinfarkt an, während sein naher Verwandter cIAP1 dies nicht tat. Beim Durchforsten vorhandener Genexpressionsdaten sah das Team, dass cIAP2 parallel zu Genen anstieg, die mit aggressiven, myeloiden Entzündungszellen assoziiert sind, was darauf hindeutet, dass cIAP2 die post-infarzielle Immunantwort verstärken könnte, statt nur Schäden widerzuspiegeln.
Weniger cIAP2, weniger Herzschaden
Um Ursache und Wirkung zu prüfen, verglichen die Forschenden normale Mäuse mit genetisch veränderten Mäusen, denen cIAP2 fehlte. Nach einem experimentellen Herzinfarkt hatten Tiere ohne cIAP2 kleinere Narben, eine bessere Pumpfunktion und weniger Flüssigkeitsansammlungen in der Lunge — alles Zeichen gesünderer Herzen. Diese Vorteile zeigten sich bei beiden Geschlechtern. Die Mikroskopie zeigte weniger sterbende Herzmuskelzellen in wichtigen Übergangsbereichen, und molekulare Analysen enthüllten Wochen später niedrigere Werte für Stress- und Umbaumarker. Im Gegensatz dazu bot das Ausschalten von cIAP1 nicht denselben Schutz und konnte in manchen Fällen die Ergebnisse sogar verschlechtern, was auf eine einzigartige, schädliche Rolle von cIAP2 in diesem Zusammenhang hinweist.
Die Milz als entzündlicher Vorratsort
Entscheidend war, wo cIAP2 aktiv war. Durch den Austausch von Knochenmark zwischen normalen Mäusen und cIAP2-defizienten Tieren zeigten die Forschenden, dass cIAP2 in blutbildenden (hämatopoetischen) Zellen einen Großteil der Schädigung verursachte. Wenn Immunzellen kein cIAP2 hatten, der Rest des Körpers aber normal war, waren die Herzen besser geschützt; der umgekehrte Tausch verschlechterte den Schaden. Bei genauerer Untersuchung der Immunorgane fanden sie, dass nach einem Herzinfarkt die Milz als Reservoir fungierte und myeloide Zellen — Neutrophile, entzündliche Monozyten und dendritische Zellen — produzierte, die dann ins Herz strömten. In Mäusen ohne cIAP2 waren diese myeloiden Milzzellen weniger zahlreich und anfälliger für Zelltod, während Lymphozyten weitgehend unbeeinflusst blieben. Signalwege, die mit Entzündungsprozessen verbunden sind, waren abgeschwächt, was darauf hindeutet, dass cIAP2 normalerweise myeloiden Zellen hilft zu überleben und weiterhin auf Gefahrenhinweise zu reagieren.
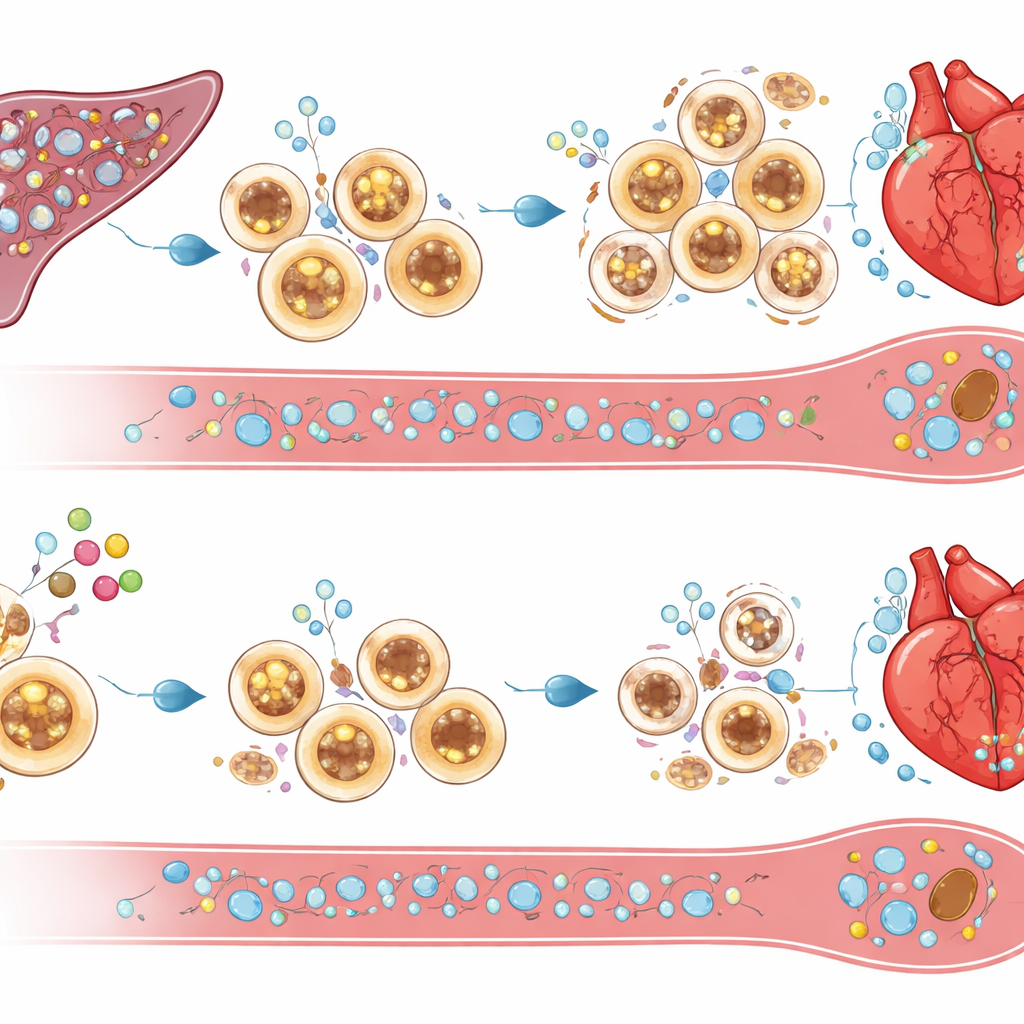
Überlebenssignale in selbstbegrenzende Aufräumarbeit verwandeln
Was tötet überschüssige Entzündungszellen, wenn cIAP2 fehlt? Die Studie deutet auf todauslösende Moleküle wie TRAIL und seinen Rezeptor DR5 sowie auf TNF-ähnliche Signale hin, die in Milz und Knochenmark von cIAP2-defizienten Mäusen nach Herzinfarkt hochreguliert waren. Das experimentelle Blockieren von TRAIL bewahrte Milzzellen vor dem Tod, stellte die starke Infiltration von Immunzellen ins Herz wieder her und beseitigte die funktionellen Vorteile des cIAP2-Verlusts. Das legt nahe, dass cIAP2 myeloide Zellen normalerweise vor diesen Todeshinweisen schützt, sodass sie sich ansammeln und die Entzündung verlängern. Ohne cIAP2 kürzen dieselben Signale das Milzreservoir zurück und verringern die Zufuhr aggressiver Zellen, die sonst das verletzte Herz überschwemmt hätten.
Den Schalter medikamentös angehen für künftige Therapien
Wichtig ist, dass das Team zeigte, dass dieser Weg mit einer bestehenden Klasse kleiner Moleküle, so genannten Smac-Mimetika, die derzeit in der Krebsforschung untersucht werden, gezielt angegangen werden kann. Mit der Verbindung LCL161 lösten sie selektiv den Abbau von cIAP-Proteinen in Milz-Immunzellen kurz nach einem Herzinfarkt aus, ohne schützende Proteine im Herzgewebe selbst zu entleeren. Behandelte Mäuse hatten weniger zirkulierende Entzündungszellen, kleinere Narben, bessere Herzfunktion und eine verbesserte Überlebensrate im Vergleich zu unbehandelten Tieren. Eine einzelne Niedrigdosis, die einen Tag nach Herzinfarkt verabreicht wurde, reichte aus, um einen kontrollierten Zelltod myeloider Milzzellen zu induzieren, lokal die TRAIL-Spiegel zu erhöhen und die Herzentzündung zu reduzieren, während sich die Gesamtheit der Immunzellen binnen vier Wochen wieder erholte. Zusammengenommen positionieren diese Ergebnisse cIAP2 als zentralen Überlebensfaktor für entzündliche Zellen nach Herzverletzung und legen nahe, dass eine kurzfristige, gezielte Hemmung von cIAP2 einen neuen, immuntherapieartigen Ansatz bieten könnte, um Herzversagen nach einem Herzinfarkt zu verhindern.
Zitation: Smyth, D., Zhang, L., Al-Khalaf, M. et al. Hematopoietic expression of cIAP2 drives inflammation and heart failure after myocardial infarction. Nat Cardiovasc Res 5, 246–261 (2026). https://doi.org/10.1038/s44161-026-00782-x
Schlüsselwörter: Myokardinfarkt, Entzündung, Immunzellen, Herzversagen, Smac-Mimetikum