Clear Sky Science · de
Integrative genomische und literaturbasierte Bewertung der desmoglein‑2‑assoziierten arrhythmogenen Kardiomyopathie mit Validierung an einer italienischen Kohorte
Warum dieses Herzgen für Familien wichtig ist
Viele plötzliche Herzprobleme bei jungen und ansonsten gesunden Menschen sind nicht zufällig – sie sind zumindest teilweise in ihrer DNA verankert. Dieser Artikel untersucht ein zentrales „Klebe“-Protein des Herzens namens Desmoglein‑2 und zeigt, wie kleine Veränderungen im entsprechenden Gen den Herzmuskel schwächen, den elektrischen Rhythmus stören und das Risiko gefährlicher Ereignisse erhöhen können. Durch die Kombination großer genetischer Datenbanken mit einer sorgfältig begleiteten italienischen Patientengruppe liefern die Forschenden klarere Antworten für Familien, die wissen möchten, was ein Testergebnis für dieses Gen wirklich bedeutet.
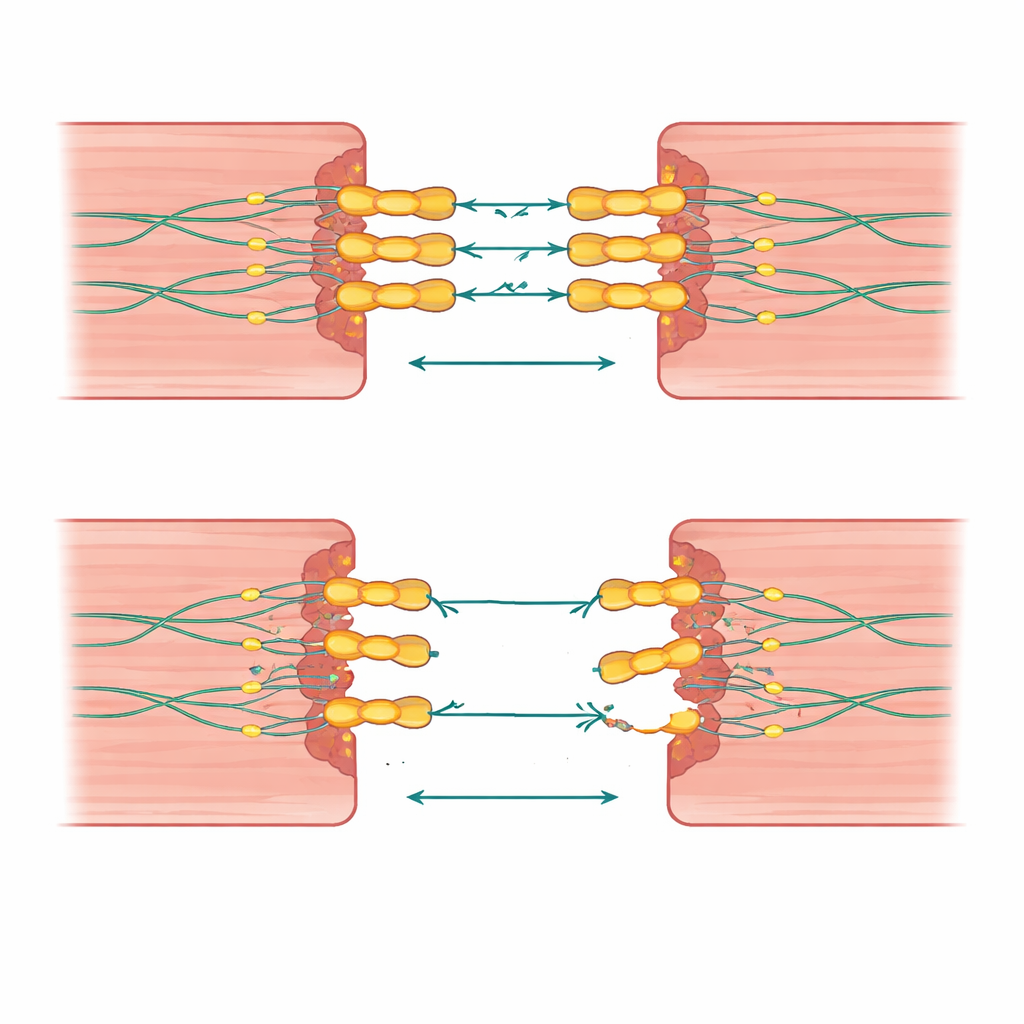
Der mechanische Klebstoff des Herzens
Herzmuskelzellen müssen beim Schlagen Millionen von Malen im Leben fest aneinander haften. Desmoglein‑2 ist Teil einer mikroskopischen, nietenähnlichen Struktur, die benachbarte Zellen miteinander verriegelt, damit sie als Team ziehen können. Die Autorinnen und Autoren erklären, wie dieses Protein von der Außenseite der Zelle, wo es an ein passendes Gegenstück der Nachbarzelle greift, bis zur Innenseite reicht, wo es in ein tragendes Gerüst einhakt. Da Desmoglein‑2 das einzige Familienmitglied ist, das in Herzmuskelzellen vorkommt, kann ein schwerer Schaden nicht durch ein Ersatzprotein kompensiert werden, wodurch das Herz besonders verwundbar wird.
Sinnvolle Genveränderungen von Hintergrundrauschen trennen
Moderne Sequenzierung entdeckt Tausende von Unterschieden im Desmoglein‑2‑Gen in der Bevölkerung, doch die meisten führen nicht zur Krankheit. Das Team sichtete systematisch 115 publizierte Studien und nutzte zwei große öffentliche Datenbanken, die zusammen mehr als 5.000 Varianten auflisteten. Mit allgemein anerkannten Regeln der medizinischen Genetik klassifizierten sie jede Veränderung neu nach ihrer Wahrscheinlichkeit, schädlich zu sein. Sie fanden, dass wirklich schädliche Varianten sich in bestimmten Regionen des Proteins anreichern – insbesondere in den äußeren Segmenten, die Calcium benötigen, um eine feste Brücke zwischen Zellen zu bilden, in einem kurzen Abschnitt, der für die Reifung des Proteins geschnitten werden muss, und in der inneren Region, die an ein anderes wichtiges Herzprotein andockt. Viele andere Veränderungen blieben „unsicher“, doch eine Untergruppe zeigte starke Hinweise auf klinische Relevanz und wurde für eine genauere Nachverfolgung markiert.
Was die italienische Patientengruppe zeigt
Um zu sehen, wie sich diese genetischen Muster bei realen Menschen auswirken, untersuchten die Forschenden 95 Personen in Italien, die Desmoglein‑2‑Varianten trugen und mit Bildgebung, Herzrhythmustests und Langzeitbeobachtung eingehend evaluiert wurden. Etwa die Hälfte erfüllte strenge Kriterien für eine arrhythmogene Kardiomyopathie, eine Erkrankung, bei der Teile des Herzmuskels allmählich durch Narben- und Fettgewebe ersetzt werden und so gefährliche Rhythmusstörungen begünstigen. Unter Verwandten, die eine Variante trugen, zeigten nur etwa vier von zehn tatsächlich Krankheitszeichen, was unterstreicht, dass ein positiver Gentest nicht zwangsläufig Krankheit bedeutet, wohl aber eine sorgfältige Überwachung erforderlich macht. Bei Betroffenen mit klarer Erkrankung gab es eine spürbare Last an schweren Rhythmuserkrankungen, während Transplantationen und Todesfälle seltener, aber dennoch vorhanden waren.
Wenn ein Treffer nicht ausreicht
Eine auffällige Erkenntnis dieser Arbeit ist, dass Anzahl und Kombination von Genveränderungen eine Rolle spielen. Personen, die zwei fehlerhafte Kopien von Desmoglein‑2 erbten, oder eine Desmoglein‑2‑Variante plus eine Veränderung in einem verwandten Herz‑Klebe‑Gen, erkrankten tendenziell früher und zeigten ausgeprägtere Schäden an beiden Herzseiten. Einige Familien trugen große Deletionen oder Duplikationen, die nicht nur Desmoglein‑2, sondern auch benachbarte Gene entfernten oder verdoppelten; auch diese Veränderungen wurden mit aggressiver Krankheit und Häufungen von plötzlichen Todesfällen in Verbindung gebracht. Im Gegensatz dazu hatten viele Verwandte mit nur einer einzelnen Veränderung milde oder keine Symptome, was darauf hindeutet, dass Hintergrundgene und Lebensfaktoren wie körperliche Belastung die Balance zwischen stillem Risiko und offenkundiger Krankheit verschieben können.
Von der Proteinform zum Patientenrisiko
Um den DNA-Code mit physischen Effekten zu verknüpfen, nutzte das Team fortgeschrittene 3D‑Proteinmodelle, um zu sehen, wie spezifische Substitutionen das Desmoglein‑2‑Gerüst lockern könnten. Veränderungen, die Calcium‑bindende Schleifen verzerrten oder wichtige Bindepunkte zerstörten, wurden als potenziell destabilisiert vorhergesagt und damit als schwächend für die Zell‑zu‑Zell‑Adhäsion eingestuft. Diese strukturellen Hinweise flossen in das Klassifizierungssystem ein und halfen, einige grenzwertige Varianten eher als wahrscheinlich schädlich oder eher als wahrscheinlich harmlos einzuordnen. Diese Verbindung zwischen molekularem Modellieren und klinischen Daten führt genetische Tests über das bloße Ablesen des Codes hinaus zu einem funktionalerem Verständnis.
Was das für Patientinnen, Patienten und Familien bedeutet
Für Familien, die von arrhythmogener Kardiomyopathie betroffen sind, bietet diese Studie sowohl Vorsicht als auch Orientierung. Sie zeigt, dass nicht jede Desmoglein‑2‑Variante ein Urteil über schwere Herzerkrankung ist, aber dass bestimmte Muster – besonders multiple Treffer oder Veränderungen in kritischen Proteinregionen – mit früheren und schwereren Problemen verbunden sind. Die Autorinnen und Autoren argumentieren, dass Personen mit diesen Varianten nicht als „gesund, bis das Gegenteil bewiesen ist“ abgetan werden sollten, sondern lebenslang mit individuell angepassten Rhythmuskontrollen und Bildgebung überwacht werden sollten. Ihr integrativer Ansatz – die Verknüpfung von Big‑Data‑Genetik, detaillierten Familienstudien und Proteinstruktur – weist den Weg zu präziseren Risikoeinschätzungen und zu sichererem Beratungsgespräch, wenn bei einem Gentest eine Veränderung in Desmoglein‑2 auftaucht.
Zitation: Pinci, S., Celeghin, R., Martini, M. et al. Integrative genomic and literature assessment of desmoglein 2-related arrhythmogenic cardiomyopathy with Italian cohort validation. Commun Med 6, 145 (2026). https://doi.org/10.1038/s43856-026-01416-w
Schlüsselwörter: arrhythmogene Kardiomyopathie, Desmoglein‑2, vererbte Herzerkrankung, genetisches Risiko, plötzlicher Herztod