Clear Sky Science · de
Bedingte Diffusion mit lokalitätsbewusster Modal-Ausrichtung zur Erzeugung diverser Proteinkonformationsensemble
Warum Proteinbewegung wichtig ist
Proteine in unseren Zellen sind keine starren Skulpturen; sie verhalten sich eher wie winzige, flexible Maschinen, die ständig ihre Form verändern. Diese Formänderungen können steuern, wie Enzyme Reaktionen katalysieren, wie Rezeptoren auf Medikamente reagieren und wie Signale durch Zellen fließen. Dennoch zeigen die meisten vertrauten Darstellungen von Proteinen nur eine einzige „Momentaufnahme“ und übersehen das reiche Ensemble an Formen, das tatsächlich existiert. Diese Arbeit stellt Mac-Diff vor, eine Methode der künstlichen Intelligenz, die schnell viele realistische Gestalten für ein gegebenes Protein erzeugen kann, sodass Forschende nicht nur sehen, wie ein Protein aussieht, sondern wie es „atmet“ und sich bewegt.
Von einzelnen Schnappschüssen zu bewegten Ensembles
Jahrzehntelang stützten sich Forschende auf mühsame Experimente oder lang laufende Molekulardynamik-Simulationen, um Proteinbewegungen zu untersuchen — beides kann langsam und teuer sein. Durchbruchswerkzeuge wie AlphaFold2 sagen inzwischen die wahrscheinlichste 3D-Struktur eines Proteins direkt aus seiner Aminosäuresequenz voraus, liefern aber in der Regel nur eine oder wenige bevorzugte Formen. Viele Proteine, insbesondere solche, die an Signalübertragung und allosterischer Regulation beteiligt sind, belegen natürlich mehrere, locker definierte Zustände. Die Autorinnen und Autoren argumentieren, dass wir, um zu verstehen, wie solche Proteine wirklich funktionieren — und um Wirkstoffe zu entwerfen, die an weniger offensichtliche, kurzlebige Formen binden — ganze Ensembles plausibler Konformationen erzeugen müssen, nicht nur eine beste Vermutung.
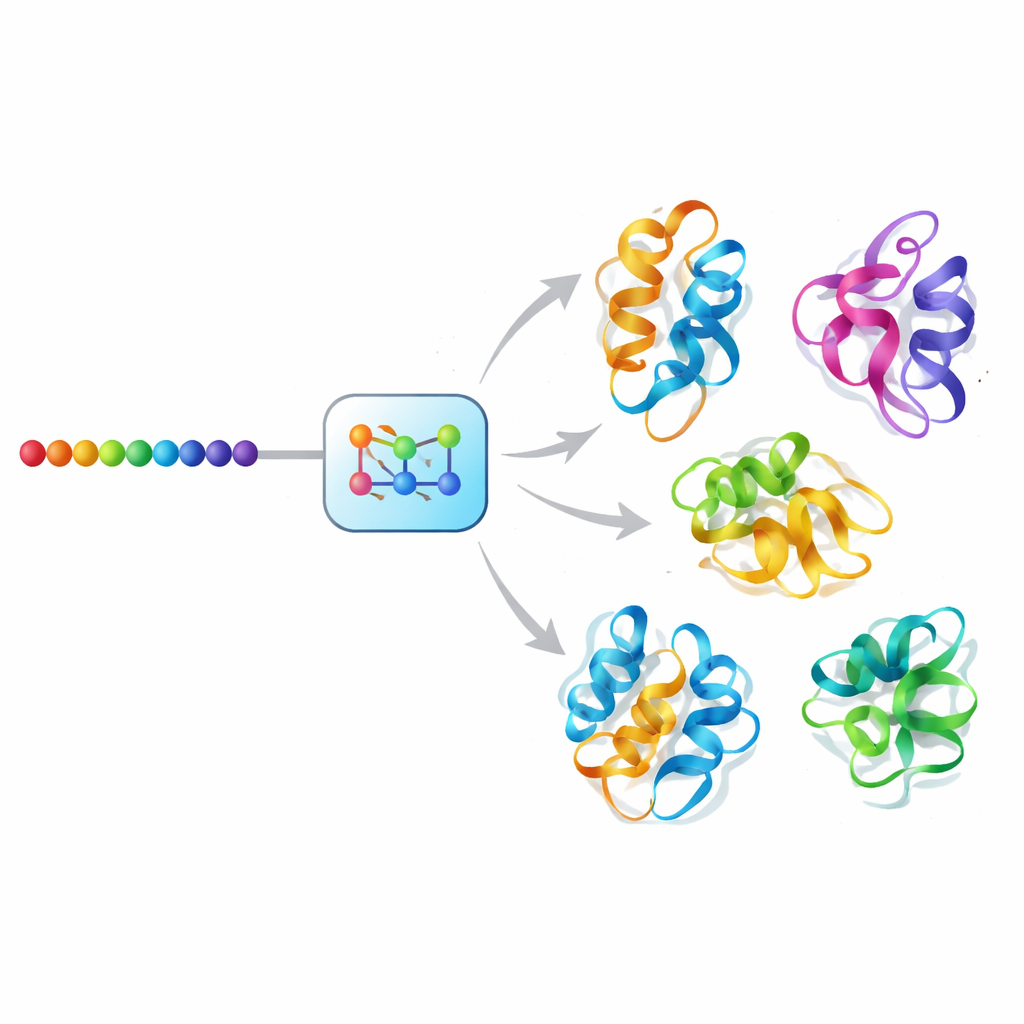
Ein KI-„Diffusions“-Ansatz für Proteinbewegung
Mac-Diff nimmt diese Herausforderung mit einem generativen Diffusionsmodell in Angriff, einer Klasse von KI, die kürzliche Fortschritte bei der Bildsynthese ermöglichte. Anstatt Fotografien zu entrauschen, entrauscht Mac-Diff abstrakte geometrische Beschreibungen von Protein-Backbones. Das Modell stellt ein Protein als Gitter paarweiser Beziehungen zwischen seinen Resten dar — Abstände und Winkel, die unempfindlich gegenüber Rotation oder Translation des gesamten Moleküls sind. In einem Vorwärts-Schritt fügt das System diesen geometrischen Mustern schrittweise Rauschen hinzu, bis sie dem Zufallsrauschen ähneln. Im Rückwärtsschritt lernt es, das Rauschen schrittweise zu entfernen, geleitet von der Aminosäuresequenz des Proteins, bis kohärente, 3D-kompatible Geometrien wieder auftauchen, die anschließend mit gängigen Strukturaufbau-Werkzeugen in vollständige Atommodelle überführt werden können.
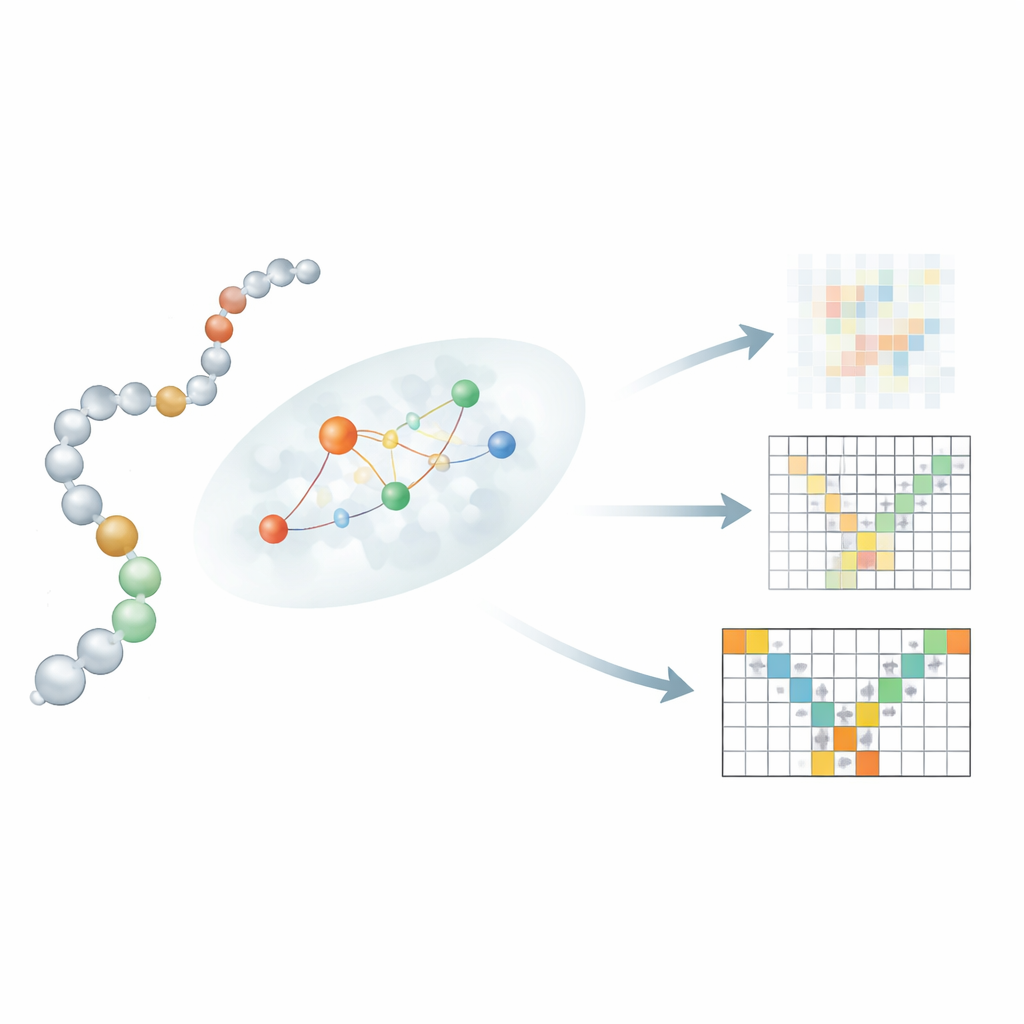
Sequenz und Struktur lokal miteinander verknüpfen
Eine zentrale Innovation liegt darin, wie Mac-Diff eine lineare Folge von Resten mit ihren 3D-Nachbarn verbindet. Einfach jedem Rest zu erlauben, jedem anderen Rest Aufmerksamkeit zu schenken, wie in Text‑zu‑Bild-Modellen, würde wichtige physikalische Einschränkungen verwischen. Stattdessen führen die Autorinnen und Autoren einen „lokalitätsbewussten“ Aufmerksamkeitsmechanismus ein, der jeden Rest auf eine kleine, wahrscheinliche Nachbarschaft von Interaktionspartnern fokussiert. Zur Abschätzung dieser Nachbarschaften nutzt Mac-Diff drei Zutaten: ein Proteinsprachemodell namens ESM-2, das den biochemischen Kontext jedes Rests kodiert; eine Kontaktkarte, die andeutet, welche Restpaare sich wahrscheinlich nahe sind; und eine einfache Regel, die Reste bevorzugt, die entlang der Kette nahe beieinanderliegen. Diese Signale werden kombiniert, sodass das Modell während des Entrauschungsprozesses bevorzugt Informationen von Resten nutzt, die physikalisch plausible Partner sind, und so seine Fähigkeit schärft, realistische, flexible Strukturen wiederherzustellen.
Tests gegen lange Simulationen und formverändernde Proteine
Die Forschenden prüften Mac-Diff auf zwei anspruchsvollen Ebenen. Zuerst fragten sie, ob es die breite Verteilung von Formen reproduzieren könne, wie sie in langen, sorgfältig berechneten Molekulardynamik-Simulationen schnell faltender Proteine und eines klassischen Benchmark-Proteins namens BPTI beobachtet wird. Über mehrere Maße, die statistische Eigenschaften der erzeugten Ensembles mit Simulationsdaten vergleichen — etwa Verteilungen von Abständen innerhalb des Proteins und die Gesamtkompaktheit — erreichte Mac-Diff vergleichbare oder bessere Ergebnisse als konkurrierende KI-Methoden und erzeugte zugleich eine größere Vielfalt an Konformationen. Es erfasste die meisten der in den Simulationen identifizierten wichtigen „metastabilen“ Zustände und reproduzierte Flexibilitätsmuster auf Rest-Ebene mit hoher Korrelation, was darauf hindeutet, dass seine Ensembles sowohl globale Faltungen als auch lokale Bewegungen realistisch widerspiegeln.
Verborgene funktionelle Zustände aufdecken
Zweitens konfrontierte das Team Mac-Diff mit Proteinen, die bekanntermaßen sehr unterschiedliche Formen bei ihrer Funktion annehmen, darunter das Enzym Adenylatkinase, das während des Energiestoffwechsels zwischen offenen und geschlossenen Formen wechselt, sowie eine kuratierte Menge von 40 Proteinen mit jeweils zwei experimentell bestimmten Konformationen. Mac-Diff erzeugte pro Protein nur 100 Kandidatenstrukturen — deutlich weniger als typische Simulationsverläufe — und fand dennoch die meisten der bekannten Zustände mit guter geometrischer Übereinstimmung. Bei der Adenylatkinase etwa lieferte es sowohl offene als auch geschlossene Konformationen mit hoher Ähnlichkeit zu Kristallstrukturen, während mehrere populäre Methoden dazu tendierten, nur einen Zustand zu bevorzugen. Das Modell lief außerdem etwa tausendmal schneller als konventionelle Simulationen auf vergleichbarer Hardware, was die systematische Erforschung von Formvielfalt deutlich praktikabler macht.
Was das für Biologie und Medizin bedeutet
Anschaulich verwandelt Mac-Diff die Sequenz eines Proteins in eine Galerie plausibler Posen statt in ein einzelnes Porträt und berücksichtigt dabei, welche Teile sich wahrscheinlich in 3D annähern oder einhaken. Indem die Methode diese Ensembles genau und effizient sampelt, bietet sie einen Weg, zu untersuchen, wie subtile Formverschiebungen Funktion zugrunde liegen, seltene aber wichtige Konformationen zu entdecken und nach Wirkstoffbindungsstellen zu suchen, die nur in transienten Zuständen auftreten. Zwar erfasst sie noch nicht die vollständig zeitlich geordneten Filme, die Simulationen liefern, doch macht Mac-Diff die dynamische Landschaft von Proteinen für viele Systeme zugänglich und verspricht neue Einsichten in Strukturbiologie, Wirkstoffdesign und Proteinengineering.
Zitation: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
Schlüsselwörter: Proteindynamik, Diffusionsmodelle, Konformationsensemble, allosterische Proteine, Arzneimittelentdeckung