Clear Sky Science · de
PFKM steuert metabolische Verschiebungen während der Differenzierung von Skelettmuskeln
Warum Muskelzellen kluge Zucker‑Nutzung brauchen
Wenn wir trainieren oder einfach vom Stuhl aufstehen, setzen unsere Skelettmuskeln Energie frei, indem sie Zucker verbrennen, um jede Kontraktion zu ermöglichen. Junge Muskelstammzellen und ausgereifte Muskelfasern gehen jedoch unterschiedlich mit Zucker um. Diese Studie beschreibt ein eingebautes Umschaltsystem, das um ein einzelnes Enzym namens PFKM zentriert ist und entscheidet, ob Glukose für unmittelbare Energie verbrannt oder umgeleitet wird, um Zellen zu schützen und zu reparieren. Das Verständnis dieses Schalters könnte neue Wege zur Behandlung von Muskelschwäche, altersbedingtem Muskelabbau und erblichen Stoffwechselkrankheiten eröffnen.
Ein Verkehrszeichen für zellulären Treibstoff
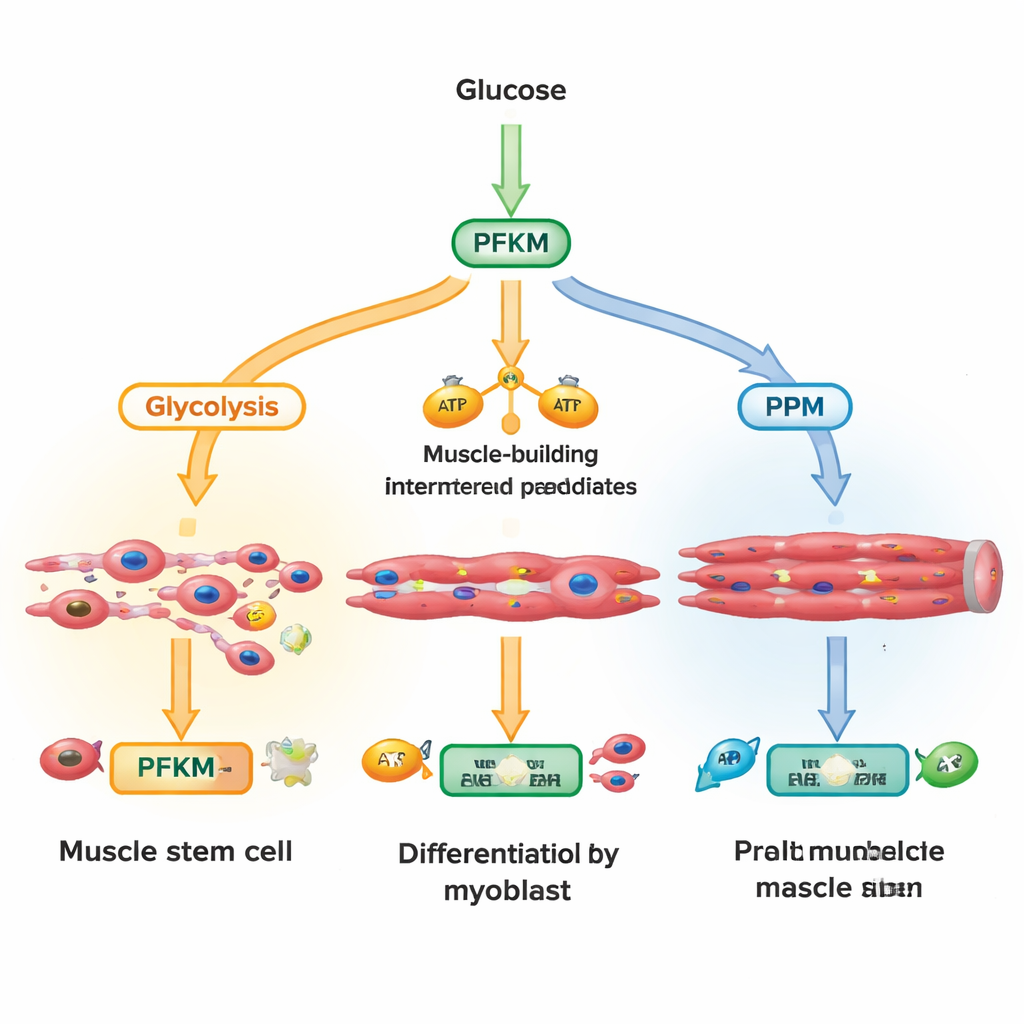
In die Zelle eindringende Glukose kann mehrere Wege gehen. Eine wichtige Route, die Glykolyse, wandelt Zucker schnell in Energie und Bausteine für wachsende Muskelfasern um. Ein anderer Weg, der Pentosephosphatweg, erzeugt Moleküle, die Zellen gegen schädlichen oxidativen Stress schützen und die Synthese von DNA und Lipiden unterstützen. Die Forschenden konzentrierten sich auf PFKM, eine Isoform des Enzyms Phosphofructokinase‑1, das an einer Schlüsselkreuzung der Glykolyse sitzt. Durch Messung von Hunderten Metaboliten über Minuten bis Stunden nach Aktivierung eines Wachstums‑Signals namens Wnt stellten sie fest, dass frühe Glykolyse‑Zwischenprodukte schnell anstiegen, während das Produkt von PFKM sank und Metaboliten des Pentosephosphatwegs zunahmen. Das deutet darauf hin, dass die Zellen PFKM aktiv drosseln, um Zucker in schützende Stoffwechselwege umzuleiten statt in reine Energieproduktion.
Das Torhüter‑Enzym markieren und entfernen
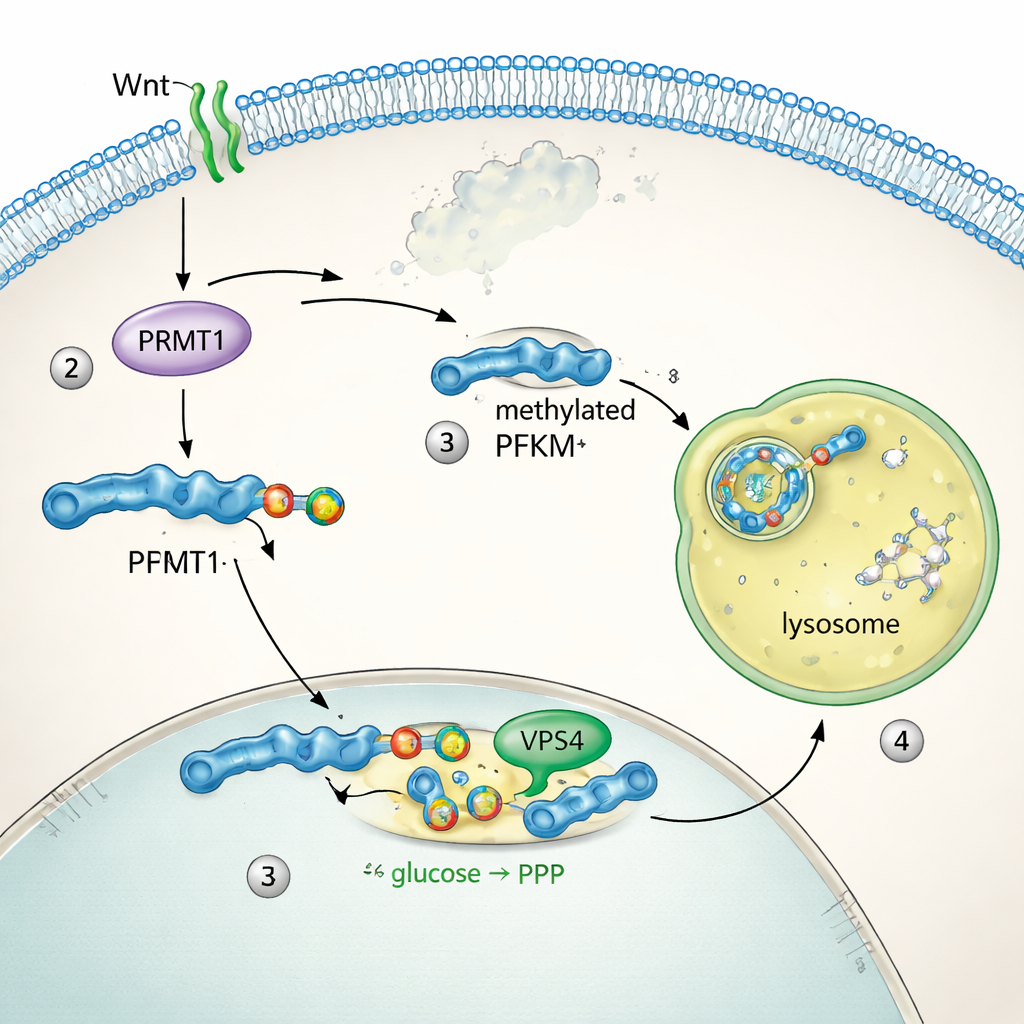
Um zu verstehen, wie PFKM kontrolliert wird, verfolgte das Team, wo das Protein innerhalb der Zellen lokalisiert ist. Im Ruhezustand verteilte sich PFKM im Cytoplasma. Innerhalb von Minuten nach Wnt‑Stimulation sammelte es sich in hellen Punkten, die mit Lysosomen, den proteinzerstörenden Kompartimenten der Zelle, überlappten. Zwei verwandte Isoformen, PFKL und PFKP, bewegten sich nicht und änderten ihre Menge nicht, was zeigt, dass nur die muskeltypische PFKM zielgerichtet wurde. Biochemische Tests zeigten, dass das Blockieren der Lysosomenfunktion den PFKM‑Abbau verhinderte, während die Hemmung des Proteasoms—des anderen Hauptsystems zum Proteinabbau—keine Wirkung hatte. Sequenzanalyse enthüllte ein kurzes, für PFKM einzigartiges „methyl‑Arginin‑Degron“‑Motif. Das Enzym PRMT1 fügte eine spezifische Methylierung an einem Arginin innerhalb dieses Motifs hinzu, und dieses Kennzeichen erlaubte der Mikroautophagie‑Maschinerie, einschließlich des Proteins VPS4, PFKM in Lysosomen zu ziehen und dort abzubauen. Das Ausschalten von PRMT1 oder VPS4 stabilisierte PFKM und verhinderte dessen Entfernung.
Vom Stammzell‑ zur arbeitenden Muskelfaser
Mithilfe großer humaner Einzelzell‑Datensätze kartierten die Autoren PFKM‑Level über viele Muskelzelltypen hinweg. Muskelstammzellen, die bis zur Bedarfssituation ruhen, wiesen sehr wenig PFKM auf, exprimierten aber viele Gene des Pentosephosphatwegs und lysosomale Komponenten. Sobald Zellen sich zur Muskelzelle verpflichteten und zu mehrkernigen Fasern fusionierten, stiegen PFKM‑Transkripte und Protein stark an, während Wnt‑Zielgene und lysosomale Gene zurückgingen. In kultivierten menschlichen und Maus‑Muskelzellen trieb Wnt PFKM schnell in Lysosomen in frühen, einkernigen Zellen, jedoch nicht in reifen mehrkernigen Fasern. Dieses Muster stützt ein Modell, in dem undifferenzierte Zellen PFKM niedrig halten—mittels lysosomalem Abbau—um einen schützenden Stoffwechsel zu begünstigen, und es dann wieder exprimieren, wenn sie in energiehungrige kontraktile Fasern übergehen.
Was passiert, wenn der Schalter klemmt
Um zu testen, wie wichtig PFKM für den Muskelaufbau ist, verringerten die Forschenden dessen Menge mittels RNA‑Interferenz. Zellen mit niedrigem PFKM hatten Schwierigkeiten, typische Muskelgene hochzufahren, produzierten weniger Myosin‑Protein und bildeten weniger und kleinere mehrkernige Fasern, obwohl die Gesamtzellzahl unverändert blieb. Metabolitprofiling zeigte verringerte nachgeschaltete glykolytische Zwischenprodukte und Brennstoffe des Tricarbonsäurezyklus, aber erhöhte Pentosephosphatweg‑Gene und Marker sowie eine verbesserte Resistenz gegen oxidativen Stress. Wichtig war, dass die Versorgung der Zellen mit 3‑Phosphoglycerat—einem glykolytischen Zwischenprodukt, das normalerweise stromabwärts von PFKM liegt—viele der Differenzierungsdefekte rettete. Muskelmarker und Faserbildung erholten sich, was zeigt, dass fehlende Metabolite und nicht der Verlust des PFKM‑Proteins selbst die Hauptursache waren.
Folgen für Muskelgesundheit und Reparatur
Für Nicht‑Spezialisten ist die zentrale Botschaft, dass Muskelzellen nicht einfach „mehr oder weniger Zucker verbrennen“; sie leiten ihn je nach Lebensphase gezielt zwischen Energiegewinnung und Zellschutz um. PFKM wirkt wie ein regulierbares Ventil an dieser Kreuzung. In stammzellähnlichen Zellen lenkt Wnt‑gesteuertes Markieren und lysosomaler Abbau von PFKM Glukose in einen Weg, der Zellen schützt und auf zukünftiges Wachstum vorbereitet. Wenn Zellen zu arbeitenden Fasern reifen, bauen sie PFKM‑Bestände wieder auf und verschieben den Stoffwechsel hin zu hoher Energieausgabe. Das Stören dieses Gleichgewichts, wie bei seltenen PFKM‑Mangelstörungen, entgleist die normale Muskelentwicklung. Indem die Studie den molekularen Griff an diesem Schalter offenlegt, legt sie nahe, dass künftige Therapien die Muskelregeneration feinjustieren oder Muskeln bei Krankheiten und Alter durch moderate Anpassung der PFKM‑Aktivität oder die Zuführung passender nachgeschalteter Metabolite schützen könnten.
Zitation: Campos, M., Nguyen, S.T., Kong, X. et al. PFKM governs metabolic shifts throughout skeletal muscle differentiation. Nat Metab 8, 489–505 (2026). https://doi.org/10.1038/s42255-026-01457-4
Schlüsselwörter: Differenzierung von Skelettmuskeln, Glukosestoffwechsel, Enzym PFKM, Pentosephosphatweg, Lysosomaler Proteinabbau