Clear Sky Science · de
Beispiellose Robustheit physik‑informierter atomarer Energiemodelle bei und oberhalb der Raumtemperatur
Warum das für die Alltagschemie wichtig ist
Computersimulationen sind die Arbeitspferde der modernen Chemie und Materialwissenschaft. Sie erlauben Wissenschaftlern, Moleküle im Rechner drehen, vibrieren und zusammenstoßen zu sehen, statt teure und zeitaufwendige Experimente durchzuführen. Wenn solche Simulationen jedoch auf maschinellem Lernen beruhen, können sie plötzlich „explodieren“ und unmögliche Molekülgestalten liefern – besonders bei höheren Temperaturen. Diese Studie stellt eine neue Art physik‑informierten maschinellen Lernmodells vor, das derartige Simulationen extrem lange und bis zu 1000 Kelvin laufen lassen kann, ohne zusammenzubrechen.
Von cleveren Abkürzungen zu fragilen Simulationen
Die traditionelle Quantenchemie berechnet Molekülenergien sehr genau, ist aber quälend langsam. Einfachere Kraftfelder sind schnell, aber häufig näherungsbehaftet. Maschinell gelernte Potentiale versuchen, das Beste aus beiden Welten zu verbinden: Sie lernen eine Abkürzung von Molekülgeometrie zu Energie und Kräften und nutzen diese Abkürzung, um molekulare Dynamik zu treiben. Auf dem Papier sehen viele solcher Modelle hervorragend aus und geben winzige mittlere Fehler auf Standard‑Testsets an. In der Praxis können diese Zahlen jedoch trügerisch sein. Wenn Moleküle während einer Simulation neue Konformationen erkunden – besonders bei höheren Temperaturen – werden viele Modelle außerhalb des Bereichs der Strukturen gedrängt, auf denen sie trainiert wurden. Statt die Moleküle sanft zu realistischen Formen zurückzuführen, können sie Kräfte vorhersagen, die Bindungen dehnen oder zerdrücken, bis das gesamte System unphysikalisch wird und die Simulation abstürzt.
Modelle auf quantenbasierten Bausteinen aufbauen
Die Autoren begegnen dieser Fragilität, indem sie ändern, was das Modell lernt und wie es durch physikalisches Vorwissen geleitet wird. Sie verwenden ein Framework namens FFLUX, das auf dem Interacting Quantum Atoms (IQA)‑Ansatz aufbaut. In IQA wird ein Molekül in „topologische Atome“ unterteilt, deren individuelle Energien direkt aus der Quantenmechanik bestimmt werden. Diese Atomenergien sind physikalisch sinnvoll und addieren sich zur Gesamtenergie des Moleküls. Anstatt willkürliche Ortsenergien zu lernen, lernen die neuen Gaussian‑Prozess‑Modelle diese quantenverwurzelten Atomenergien und erhalten so einen tiefen physikalischen Anker für jede Vorhersage. Vier flexible organische Moleküle – peptid‑terminiertes Glycin und Serin, Malondialdehyd und Aspirin – dienen als anspruchsvolle Testfälle wegen ihrer vielen internen Bewegungen und der bekannten Schwierigkeit für bestehende maschinelle Lern‑Kraftfelder.
Dem Modell beibringen, Ärger zu erwarten
Eine Schlüsselinnovation liegt darin, wie der Gaussian‑Prozess bereits vor dem ersten Datensatz eingerichtet wird: seine „Mittelwertfunktion“, die kodiert, was das Modell in schlecht bekannten Regionen annimmt. Die meisten früheren Arbeiten setzen diesen Mittelwert einfach auf null und tun damit so, als habe das Modell keine Vorannahmen. Die Autoren verschieben diesen Mittelwert stattdessen gezielt in Richtung höherer Atomenergiezustände, wobei er physikalisch sinnvoll bleibt. Diese Designentscheidung bewirkt, dass das Modell bei notwendiger Extrapolation – etwa wenn Bindungen zeitweise überdehnt sind – naturgemäß Vorhersagen bevorzugt, die extreme Verzerrungen bestrafen. In umfangreichen Tests verhielten sich Modellvarianten, die sich nur in diesem Vorwissen unterschieden, sehr unterschiedlich. Modelle mit konventionellen oder auf niedrige Energie ausgerichteten Mittelwerten überlebten oft weniger als eine Pikosekunde, bevor ein Molekül explodierte oder kollabierte. Im Gegensatz dazu ergab der beste hochenergetische Mittelwert (MF5 genannt) Simulationen, die über das volle Nanosekunden‑Testfenster bei Temperaturen von 300 bis 1000 Kelvin für alle vier Moleküle stabil blieben.
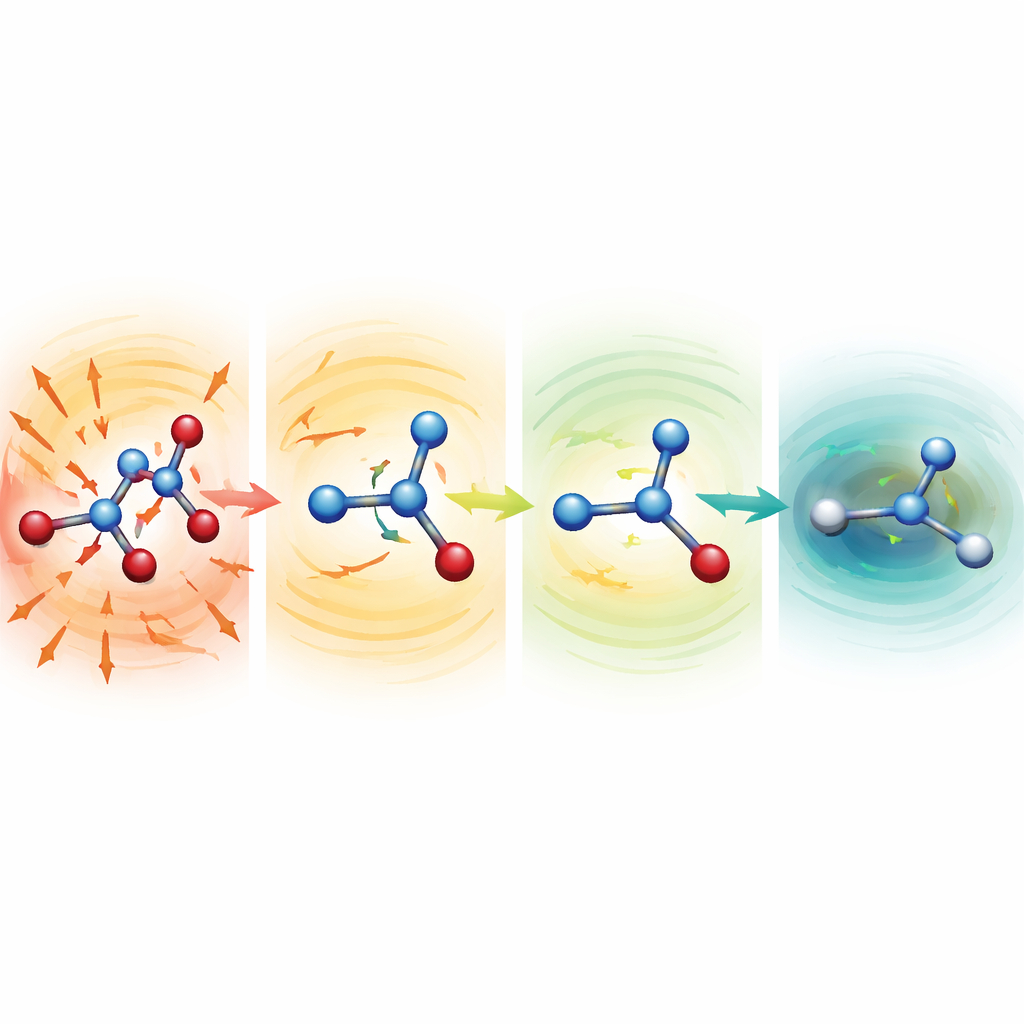
Zusehen, wie verzerrte Moleküle sich selbst heilen
Um zu untersuchen, warum die robusten Modelle so gut funktionieren, starteten die Forschenden Simulationen von absichtlich verstümmelten Strukturen mit stark gedehnten oder komprimierten Bindungen. Bei Serin, Aspirin und Malondialdehyd lagen diese Ausgangspunkte hunderte bis mehr als tausend Kilokalorien pro Mol über einer normalen Struktur – Konfigurationen, die normalerweise katastrophal wären. Mit schwächeren Mittelwertfunktionen flogen die Moleküle schnell auseinander. Mit der MF5‑Konfiguration zeigten die vorhergesagten Kräfte jedoch sofort in Richtungen, die gedehnte Bindungen verkürzten und gequetschte Bindungen verlängerten. Innerhalb von einigen Dutzend bis Hunderten von Zeitschritten entspannten sich die Moleküle zu realistischen Formen und entwickelten sich dann stabil weiter. Das Team zeigte außerdem, dass dieselben Modelle, obwohl sie nie auf Kräfte trainiert wurden, Geometrie‑Optimierungen von Alanin‑Dipeptid steuern können und bekannte energiearme Konformationen sowie relative Energien innerhalb weniger Zehntel Kilokalorien pro Mol reproduzieren – und das bei etwa 200‑fach geringeren Kosten im Vergleich zu vollständigen Quantumberechnungen.
Lange, heiße Simulationen auf gewöhnlicher Hardware
Robustheit bedeutet nicht nur, einen schwierigen Start zu überstehen; sie muss sich über Millionen oder Milliarden von Zeitschritten halten. Die Autoren testeten ihre besten Modelle weiter, indem sie 50 unabhängige Simulationen bei 500 Kelvin laufen ließen, jeweils 10 Nanosekunden, für ihre vier Testmoleküle. Keine dieser Läufe stürzte ab, was eine kombinierte Simulationszeit von einer halben Mikrosekunde ergab – ungewöhnlich für hochmoderne maschinell gelernte Kraftfelder. Noch bemerkenswerter ist, dass die Simulationen effizient auf Standard‑CPUs liefen und pro Zeitschritt manchen prominenten neuronalen Netzpotenzialen ebenbürtig waren oder diese übertrafen, die leistungsfähige GPUs benötigen. Währenddessen erkundeten die Moleküle vielfältige Gestalten und metastabile Zustände, was zeigt, dass Robustheit nicht durch künstliches Einfrieren der Bewegung oder Erzwingen starrer Strukturen erreicht wurde.
Was das für die zukünftige molekulare Modellierung bedeutet
Für Nicht‑Experten ist die Kernbotschaft: Nicht alle Modelle mit niedrigen Fehlern sind vertrauenswürdig, wenn sie stark beansprucht werden. Indem die Autoren ihre Modelle in quantenabgeleiteten Atomenergien verankerten und die eingebauten Erwartungen des Modells sorgfältig in Richtung hoher Energiezustände abstimmten, schufen sie eine Familie von Potentialen, die natürlicherweise „rückführende Kräfte“ liefern – das molekulare Äquivalent eines Sicherheitsgurts –, die Simulationen selbst bei hohen Temperaturen und verzerrten Anfangszuständen physikalisch halten. Dieser Ansatz verspricht zuverlässigere und längere Simulationen komplexer Moleküle und weist den Weg zu künftigen Erweiterungen, bei denen ähnliche physik‑informierte Modelle kondensierte Phasen und feine Wechselwirkungen wie Dispersionskräfte behandeln, während sie rechnerisch praktikabel bleiben.
Zitation: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Schlüsselwörter: maschinelle Lern‑Kraftfelder, Robustheit molekularer Dynamik, Gaussian‑Prozess‑Potenziale, physik‑informierte Modellierung, quantenbasierte Atomenergien