Clear Sky Science · de
Benchmarking von Deep-Learning-Modellen zur Vorhersage der Wirksamkeit von Krebsmedikamenten (IC50) mit Einsichten für Pharmazeutiker
Warum diese Forschung für zukünftige Krebsmedikamente wichtig ist
Die Entwicklung neuer Krebsmedikamente ist langsam und teuer, weil jede vielversprechende Verbindung in lebenden Zellen getestet werden muss, um zu sehen, wie stark sie deren Wachstum hemmt. Diese Studie stellt eine pragmatische Frage: Können moderne Werkzeuge der künstlichen Intelligenz diese Testergebnisse zuverlässig im Voraus vorhersagen und so Zeit und Kosten im Labor sparen? Die Autorinnen und Autoren vergleichen systematisch mehrere verbreitete Deep-Learning-Systeme, untersuchen, wann sie erfolgreich oder fehleranfällig sind, und schlagen sogar eine realistischere Methode vor, um ihren Nutzen für praktizierende Pharmazeutiker einzuschätzen.
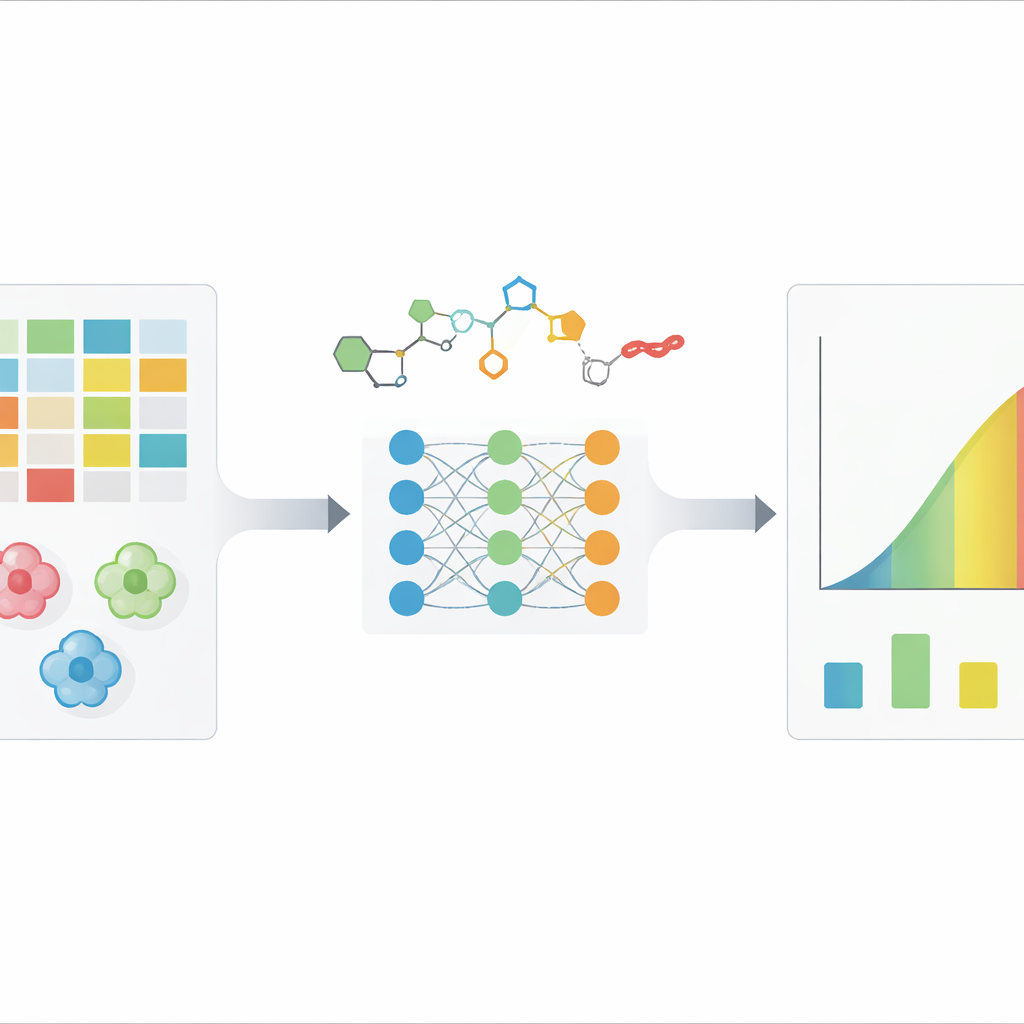
Wie stark ein Wirkstoff Krebszellen bekämpft – Messgrößen
Wenn Forschende eine potenzielle krebshemmende Verbindung testen, berichten sie häufig eine Zahl namens IC50: die Konzentration, bei der der Wirkstoff das Zellwachstum um die Hälfte reduziert. Ein niedriger IC50-Wert bedeutet ein potentes Mittel. Dieselbe Verbindung kann jedoch in verschiedenen Krebszelllinien sehr unterschiedliche IC50-Werte zeigen, und selbst wiederholte Tests derselben Wirkstoff–Zell-Kombination können je nach Assay und Bedingungen um das Mehrfache variieren. Traditionelle rechnergestützte Designmethoden erfassen, wie ein Molekül zu einem einzelnen Proteinziel passt, haben aber Schwierigkeiten mit der gesamten Komplexität lebender Zellen. Neuere Deep-Learning-Methoden versuchen, Muster direkt aus großen Datensätzen zu lernen, die chemische Strukturen und detaillierte genetische Informationen über Krebszellen mit ihren gemessenen IC50-Werten verknüpfen.
Fünf Deep-Learning-Tools im Test
Die Autorinnen und Autoren untersuchten fünf führende Deep-Learning-Modelle, die jeweils eine andere Strategie zur Darstellung sowohl von Wirkstoffen als auch von Krebszellen verwenden. Manche behandeln Moleküle als Graphen aus Atomen und Bindungen; andere transformieren Zellgenetik in strukturierte Netzwerke biologischer Prozesse oder heben die informativsten Gene hervor. Alle Modelle wurden auf denselben kuratierten Daten aus einer großen Ressource namens GDSC trainiert und bewertet, die Zehntausende gemessener IC50-Werte enthält. Das Team baute außerdem eine bewusst einfache Vergleichsmethode: eine „Baseline“, die Biologie und Chemie ignoriert und lediglich durchschnittliche IC50-Werte aus den Trainingsdaten vorhersagt. So konnten sie nicht nur fragen, welches Deep-Modell am besten ist, sondern auch, ob eines von ihnen tatsächlich einen sehr naiven Shortcut übertrifft.
Eine realistischere Methode zur Bewertung von Vorhersagen
Gängige Machine-Learning-Metriken wie Korrelation und Root-Mean-Squared Error können beeindruckend wirken, sind für experimentell arbeitende Wissenschaftlerinnen und Wissenschaftler aber oft schwer zu interpretieren. Um diese Lücke zu schließen, formten die Autorinnen und Autoren die Vorhersagequalität in intuitivere Maße um, etwa Prozentfehler und Fehler auf einer logarithmischen Skala, die direkt den Mehrfachunterschieden im IC50 entspricht. Entscheidend quantifizierten sie auch, wie verrauscht reale IC50-Messungen sind, indem sie eine große Bioaktivitätsdatenbank durchsuchten. Sie zeigten, dass unter häufigen Assay-Bedingungen 90 % wiederholter IC50-Messungen für dasselbe Wirkstoff–Zell-Paar innerhalb von etwa einem siebenfachen Bereich liegen. Darauf aufbauend definierten sie eine neue Metrik, Experimental Variability-Aware Prediction Accuracy (EVAPA): den Prozentsatz der Modellvorhersagen, die innerhalb dieses experimentell realistischen Bereichs liegen.
Wo die Modelle glänzen und wo sie Probleme haben
Wenn die Daten zufällig aufgeteilt wurden, sodass viele Wirkstoffe und Zelllinien sowohl im Trainings- als auch im Testset auftauchten, schnitten alle Deep-Learning-Modelle gut ab. Sie zeigten starke Korrelationen mit gemessenen IC50-Werten und hohe EVAPA-Werte und übertrafen klar die einfache Baseline. Die Leistung blieb relativ gut, als die Modelle generalisieren sollten auf völlig neue Zelllinien, während ihnen vertraute Wirkstoffe weiterhin bekannt waren; in diesem Fall schnitt die Baseline überraschend gut ab, was darauf hindeutet, dass das durchschnittliche Verhalten eines Wirkstoffs über viele Zelltypen bereits nützliche Informationen enthält. Die eigentlichen Probleme traten auf, wenn die Modelle mit neuen chemischen Strukturen konfrontiert wurden: Die Genauigkeit fiel stark ab, Korrelationen näherten sich null oder wurden sogar negativ, und in einigen Tests glich oder übertraf die einfache Baseline die Deep-Modelle. Das Team prüfte auch, ob Vorhersagefehler von grundlegenden Wirkstoffeigenschaften wie Größe, Polarität oder Flexibilität oder vom Gewebeursprung der Zelllinien abhängen. Sie fanden nur schwache Zusammenhänge, was nahelegt, dass die Modelle über verschiedene Chemien und Krebsarten hinweg etwa gleich gut funktionieren — aber bei wirklich neuartigen Verbindungen versagen.
Wirklich neue Moleküle aus aktuellen Studien testen
Um über öffentliche Datenbanken hinauszugehen, sammelten die Autorinnen und Autoren mehr als 150 kürzlich berichtete krebshemmende Verbindungen aus der medizinisch‑chemischen Literatur und testeten mehrere der Deep-Learning-Modelle an diesen bislang ungesehenen Molekülen. Die Ergebnisse spiegelten das „neue Wirkstoff“-Szenario in den GDSC-Daten wider: Die Vorhersagen waren verrauscht, mit hohen Prozentfehlern und nur mäßigen Anteilen an Vorhersagen innerhalb realistischer experimenteller Grenzen. Dennoch deutete das Verhalten der Modelle über verschiedene Assay-Typen hinweg darauf hin, dass sie einige assay‑unabhängige Muster darin erfassten, wie Wirkstoffe Zellen beeinflussen. Ein einfacher Webserver, der aus diesen Modellen aufgebaut wurde, erlaubt Chemikerinnen und Chemikern nun, eine Struktur einzugeben und vorhergesagte IC50-Werte für Hunderte von Krebszelllinien zu erhalten — mit dem Vorbehalt, dass die Zuverlässigkeit am höchsten ist, wenn das neue Molekül denen im Trainingssatz ähnelt.
Was das für die Wirkstoffforschung bedeutet
Diese Arbeit zeigt, dass aktuelle Deep-Learning-Werkzeuge bereits nützlich sind, um Krebswirkstoffideen innerhalb vertrauter chemischer Territorien zu ordnen und zu erkunden, aber sie sind weit davon entfernt, Kristallkugeln für wirklich neue Moleküldesigns zu sein. Indem sie hervorheben, dass ein grobes durchschnittsbasiertes Modell manchmal mit komplexen neuronalen Netzen konkurrieren kann, und indem sie eine Genauigkeitsmessung einführen, die in realer experimenteller Variabilität verankert ist, geben die Autorinnen und Autoren Pharmazeutikerinnen und Pharmazeutikern eine klarere Vorstellung davon, was von IC50‑Vorhersagesoftware zu erwarten ist. Die Botschaft ist ausgewogen: Diese Modelle sind vielversprechende Hilfsmittel für die Wirkstoffforschung, insbesondere wenn sie sorgfältig benchmarked werden, aber bedeutende Fortschritte in Architektur und Training — insbesondere für außerhalb der Verteilungs liegende Moleküle — sind noch nötig, bevor sie zuverlässig die Suche nach der nächsten Generation von Krebstherapien anleiten können.
Zitation: Garai, U., Pal, A.S., Ghosh, K. et al. Benchmarking deep learning models for predicting anticancer drug potency (IC50) with insights for medicinal chemists. Commun Chem 9, 106 (2026). https://doi.org/10.1038/s42004-026-01916-9
Schlüsselwörter: Wirksamkeit von Krebsmedikamenten, IC50-Vorhersage, Deep-Learning-Modelle, Krebszelllinien, computergestützte Wirkstoffforschung