Clear Sky Science · de
Feinabstimmung von AlphaFold mit begrenzten Kryo‑EM‑Beobachtungen
Warum Proteine so schwer zu sehen sind
Proteine sind winzige molekulare Maschinen, die nahezu jeden Prozess in unserem Körper antreiben – von der Energiegewinnung bis zu Nervensignalen. Um zu verstehen, wie sie funktionieren und wie Medikamente sie beeinflussen können, müssen Wissenschaftler ihre exakten dreidimensionalen Formen kennen. Zwei leistungsfähige Werkzeuge haben sich dafür etabliert: Kryo‑Elektronenmikroskopie (kryo‑EM), die viele unscharfe Momentaufnahmen gefrorener Proteine erstellt, und AlphaFold, ein KI‑System, das Proteinstrukturen aus Sequenzen vorhersagt. In vielen realen Experimenten sind die kryo‑EM‑Daten jedoch unvollständig, und AlphaFolds Vorhersagen stimmen nicht immer mit der Wirklichkeit überein. Dieses Papier stellt CoCoFold vor, eine Methode, die AlphaFold beibringt, direkt auf schwierige kryo‑EM‑Daten zu hören und seine Vorhersagen entsprechend zu verbessern.
Wenn die Kamera zu wenig sieht
Kryo‑EM funktioniert, indem Proteine blitzeingefroren und enorme Mengen einzelner Partikel aus vielen Blickwinkeln abgebildet werden, um diese Aufnahmen dann zu einer 3‑D‑Karte zu kombinieren. In der Praxis haben Forscher jedoch oft nicht genügend brauchbare Bilder. Manchmal erscheint das Protein nur kurz in einem energiereichen Zustand, sodass nur sehr wenige Partikel erfasst werden. In anderen Fällen bevorzugen Proteine bestimmte Orientierungen auf der Eisoberfläche, sodass viele Betrachtungswinkel fehlen. Beide Probleme führen zu verschwommenen, unvollständigen Karten, die schwer in zuverlässige atomare Modelle zu übersetzen sind. Bestehende Software kann AlphaFold‑Vorhersagen in solche Karten einpassen, hängt dabei aber stark von scharfen, hochauflösenden Daten ab.
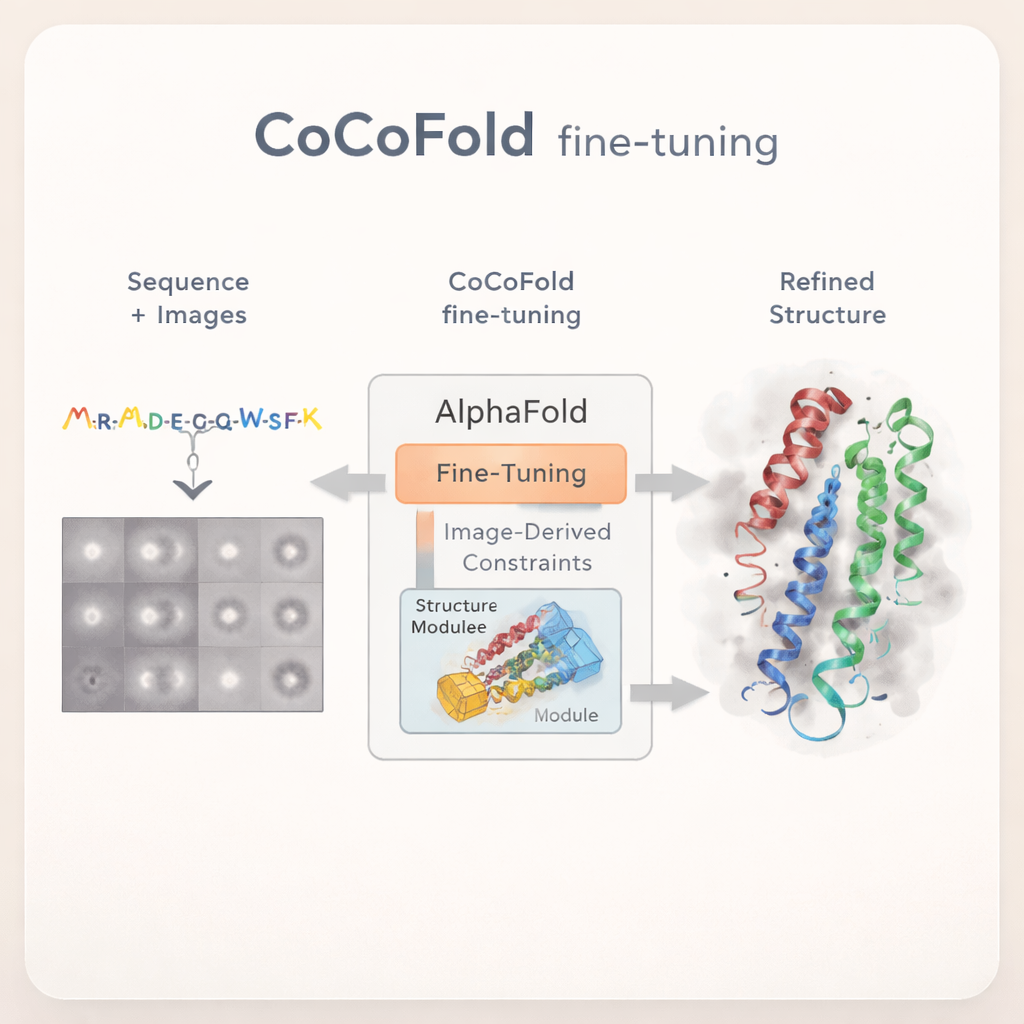
AlphaFold beibringen, aus Rohbildern zu lernen
CoCoFold verfolgt einen anderen Ansatz: Anstatt sich auf eine vollständig rekonstruierte 3‑D‑kryo‑EM‑Karte zu stützen, nutzt es direkt die rohen 2‑D‑Partikelbilder, um AlphaFold feinzujustieren. Die Methode startet von einer AlphaFold‑Multimer‑Vorhersage und hält den Großteil des ursprünglichen Netzwerks eingefroren, um dessen breites Wissen über Proteinfaltung zu bewahren. Nur der abschließende, strukturaufbauende Teil darf sich ändern. Ein leichter „Adapter“ wird hinzugefügt, um aus den kryo‑EM‑Bildern gewonnene Informationen in dieses Strukturmodul zu speisen, wodurch das Modell sanft in Richtung Geometrien gelenkt wird, die mit den experimentellen Daten kompatibel sind, ohne bekannte physikalische Prinzipien der Proteine zu verletzen.
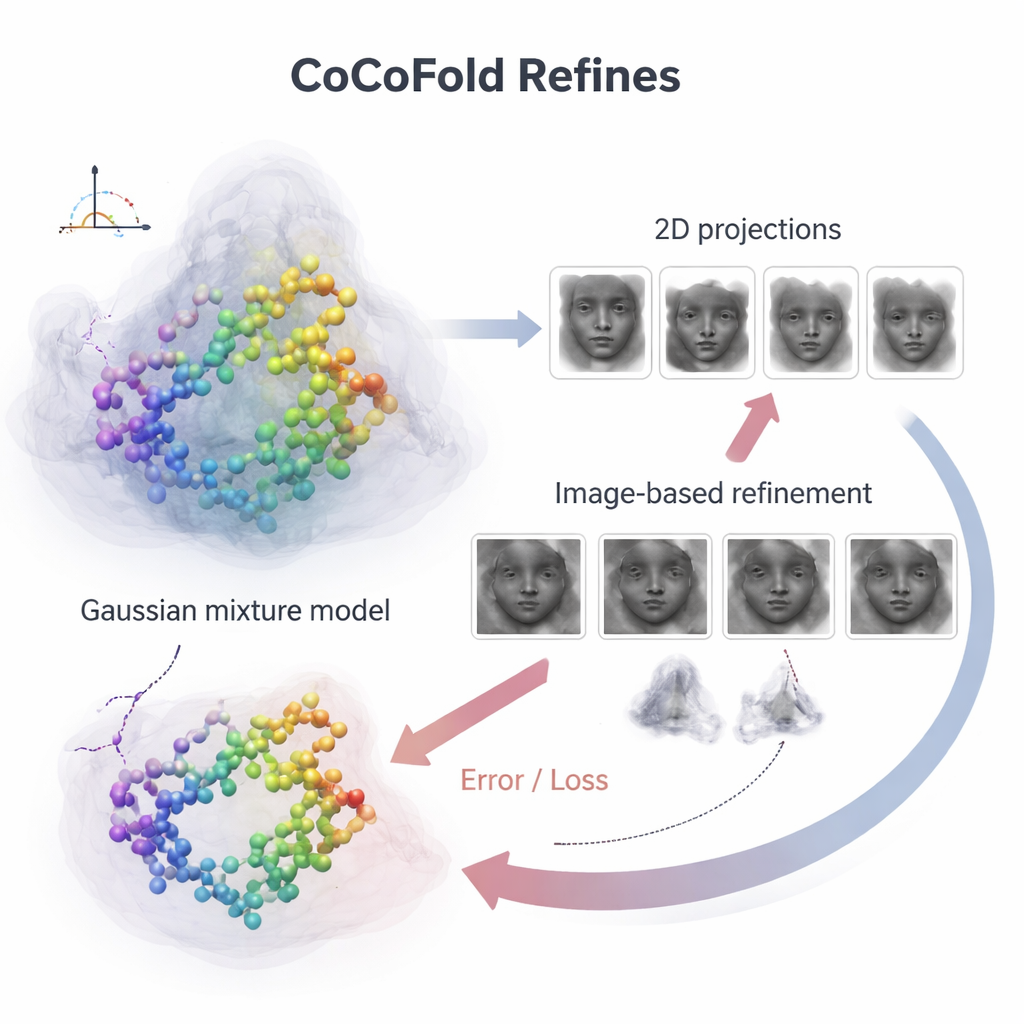
Bilder in strukturelles Feedback verwandeln
Um einzelne Proteinatome mit den verrauschten Mikroskopbildern zu verbinden, baut CoCoFold ein glattes, flexibles Abbild der vorhergesagten Struktur aus sich überlappenden dreidimensionalen Klumpen, einer sogenannten Gaußschen Mischung. Aus dieser Darstellung simuliert es, wie das Protein im Mikroskop bei denselben Blickrichtungen und Aufnahmebedingungen wie im Experiment erscheinen würde. Diese simulierten Schnappschüsse werden dann Ring für Ring im Frequenzbereich mit den tatsächlichen kryo‑EM‑Partikeln verglichen, um die Übereinstimmung zu prüfen. Jede Diskrepanz wird zu einem Feedbacksignal, das durch das Netzwerk zurückfließt und sowohl das Proteinmodell als auch die Dichtedarstellung leicht anpasst. Nach dem Training wird das atomare Modell zusätzlich mit einem physikbasierten Verfeinerungsschritt bereinigt, um lokale geometrische Kollisionen zu entfernen.
Genau bleiben, wenn Daten knapp oder verzerrt sind
Die Autoren testeten CoCoFold an mehreren experimentellen und simulierten Datensätzen, die die beiden Hauptprobleme der kryo‑EM nachahmen sollten: zu wenige Partikel und große Lücken in den Betrachtungswinkeln. Unter diesen schwierigen Bedingungen verpassten Standardwerkzeuge – einschließlich anderer tief lernender Methoden, die auf rekonstruierten Karten beruhen – oft Bereiche des Proteins, platzierten Helices falsch oder verloren feine Details, sobald die Karten unschärfer wurden. CoCoFold hingegen lieferte konsistent Modelle, die den bekannten Referenzstrukturen näher und vollständiger entsprachen. Die Fehler blieben klein, selbst wenn die Partikelanzahl drastisch reduziert oder große Kegel von Blickrichtungen fehlten, was darauf hindeutet, dass das direkte Lernen aus Rohbildern entscheidende Informationen bewahrt, die kartenbasierte Ansätze verwerfen.
Was das für die zukünftige Strukturbiologie bedeutet
Für Nicht‑Spezialisten ist die Kernbotschaft, dass CoCoFold wie ein Übersetzer zwischen leistungsfähigen KI‑Vorhersagen und unvollkommenen experimentellen Daten wirkt. Anstatt allein AlphaFold oder kryo‑EM zu vertrauen, lässt es beide Quellen gegenseitig aufeinander einwirken, insbesondere in den schwierigen Fällen, in denen Experimente nur eine partielle Sicht liefern. In einfachen Fällen mit reichlich qualitativ hochwertigen Daten funktionieren die bestehenden kartenbasierten Werkzeuge weiterhin sehr gut. Wenn Partikel jedoch selten sind oder Orientierungen fehlen – typische Situationen beim Erforschen flüchtiger oder fragiler Proteinzustände – bietet CoCoFold einen Weg, aus sonst verlorenen Informationen zuverlässige atomare Modelle zu gewinnen.
Zitation: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Schlüsselwörter: cryo-EM, AlphaFold, Proteinstruktur, Tiefes Lernen, Strukturbiologie