Clear Sky Science · de
Mitochondriale Energiekrise liegt FLVCR1-assoziierter sensorischer Neuropathie zugrunde
Wenn Schmerznerven die Energie ausgeht
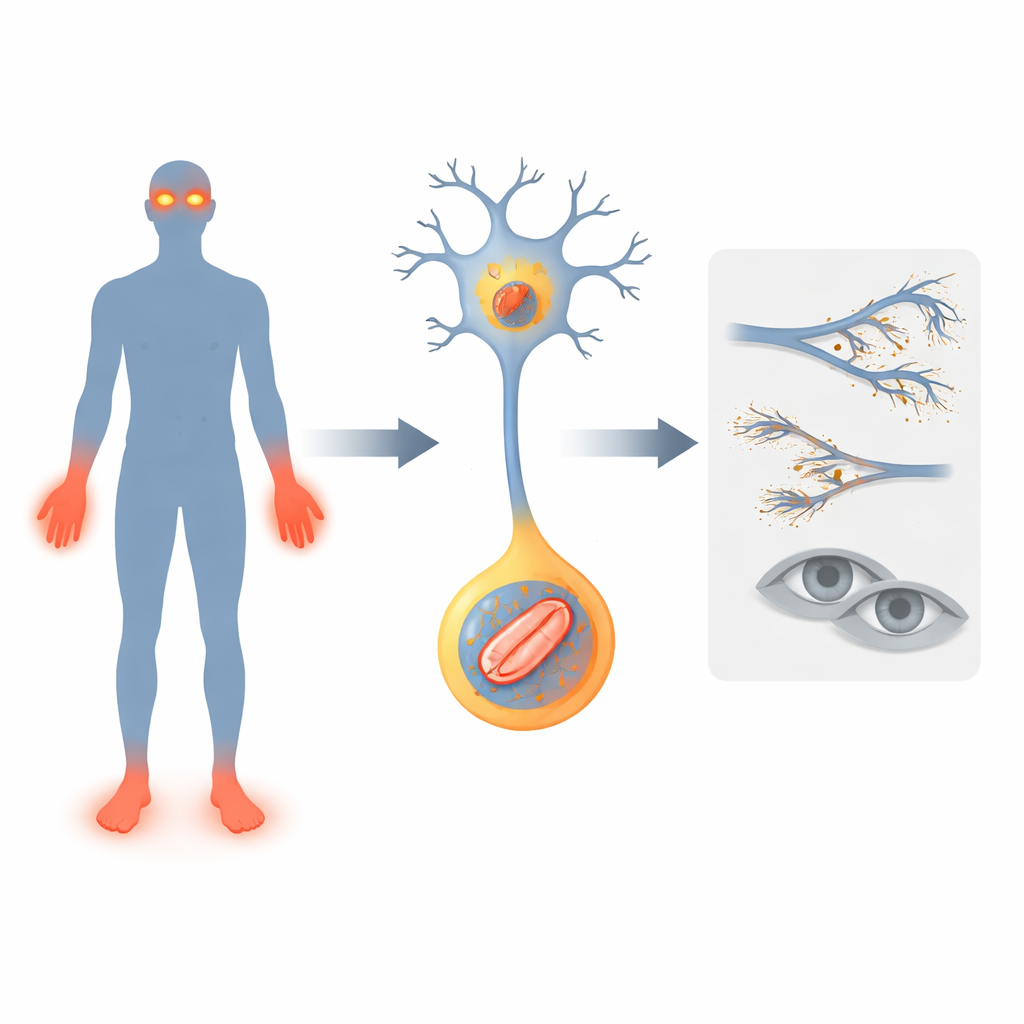
Manche Menschen werden mit nahezu fehlender Schmerzempfindung geboren. Auf den ersten Blick mag das wie ein Geschenk wirken, doch schnell wird es zum Fluch: Ohne Schmerz als Warnsignal häufen sich Verbrennungen, Brüche, Infektionen und sogar Erblindung. Diese Studie untersucht eine seltene Form solcher Schmerzverlust-Störungen und enthüllt einen überraschenden Übeltäter: winzige Kraftwerke in Nervenzellen, deren Energieproduktion massiv gestört ist.
Ein Gen, das die Alarmglocken zum Schweigen bringt
Die Forschenden konzentrieren sich auf ein Gen namens FLVCR1, das bereits mit seltenen Nervenleiden in Verbindung gebracht wurde, bei denen Betroffene das Schmerzempfinden verlieren, einen unsicheren Gang entwickeln und manchmal fortschreitenden Sehverlust erleiden. Sie beschreiben zwei neue Patientinnen bzw. Patienten mit Veränderungen in FLVCR1. Beide Kinder zeigten früh Probleme: verzögerte motorische Meilensteine, häufiges Stolpern, schwere Infektionen und Verstümmelungen an Fingern und Zehen, weil Verletzungen unbemerkt blieben. Einer der Patienten entwickelte zudem eine degenerative Augenkrankheit namens Retinitis pigmentosa, die zur Nachtblindheit führte. Diese Fälle weiten das Bild der möglichen FLVCR1-Defekte beim Menschen und stützen die Annahme, dass dieses Gen entscheidend dafür ist, Schmerz wahrnehmende Nerven und lichtempfindliche Zellen in der Netzhaut am Leben zu erhalten.
Die Krankheit in winzigen Fischen modellieren
Um zu erforschen, wie FLVCR1 die Entwicklung sensorischer Nerven beeinflusst, nutzte das Team Zebrafische, deren transparente Embryonen direkte Beobachtungen von Nervenzellen erlauben. Sie reduzierten die Expression der fischspezifischen Genvariante flvcr1a mit genetischen Werkzeugen. Fische mit reduziertem flvcr1a hatten weniger dorsale Wurzelganglien, Nervenzellcluster, die Berührung und Schmerz entlang der Wirbelsäule detektieren. Verhaltenstests zeigten, dass diese Fische sich weniger aus eigener Kraft bewegten und nach leichtem Schwanzkontakt nur kurze Strecken schwammen, was auf eine abgeschwächte sensorische Reaktion hinweist. Weil frühere Mausmodelle zu früh verstarben, um ihre sensorischen Nerven zu analysieren, liefern diese Zebrafische das erste lebende System, in dem FLVCR1-assoziierte Nervenstörungen und Verhaltensänderungen detailliert verfolgt werden können.
Drei gestörte Wege konvergieren an den Zellkraftwerken
FLVCR1 sitzt in Zellmembranen und reguliert mehrere Schlüsselsubstanzen. Frühere Arbeiten deuteten auf Rollen im Umgang mit Cholin (einem Baustein von Membranlipiden), Häm (dem eisenhaltigen Pigment, das viele Enzyme antreibt) und dem Calciumsignalfluss zwischen Zellkompartimenten hin. Die Wissenschaftler sammelten Hautzellen (Fibroblasten) von vier Patientinnen bzw. Patienten mit unterschiedlichen FLVCR1-Mutationen und verglichen sie mit Zellen gesunder Personen sowie symptomfreien Trägern. Sie fanden, dass Patientenzellen niedrigere Cholinwerte und flüssigere Zellmembranen aufwiesen — Veränderungen, die das empfindliche Lipidmilieu stören könnten, das Mitochondrien benötigen. Zudem entdeckten sie, dass ein entscheidendes Enzym zur Hämproduktion in Mitochondrien, ALAS1, weniger aktiv war, obwohl der Gesamtgehalt an Häm nahezu normal erschien. Gleichzeitig waren die Kontaktstellen zwischen endoplasmatischem Retikulum und Mitochondrien — wo normalerweise Calcium in die Mitochondrien gelangt — kürzer und seltener, und der Calcium-Einstrom in die Mitochondrien war reduziert. Drei Probleme — Cholinmangel, träge Hämproduktion und geschwächte Calciumübertragung — deuteten alle auf eine beeinträchtigte mitochondriale Leistungsfähigkeit hin.
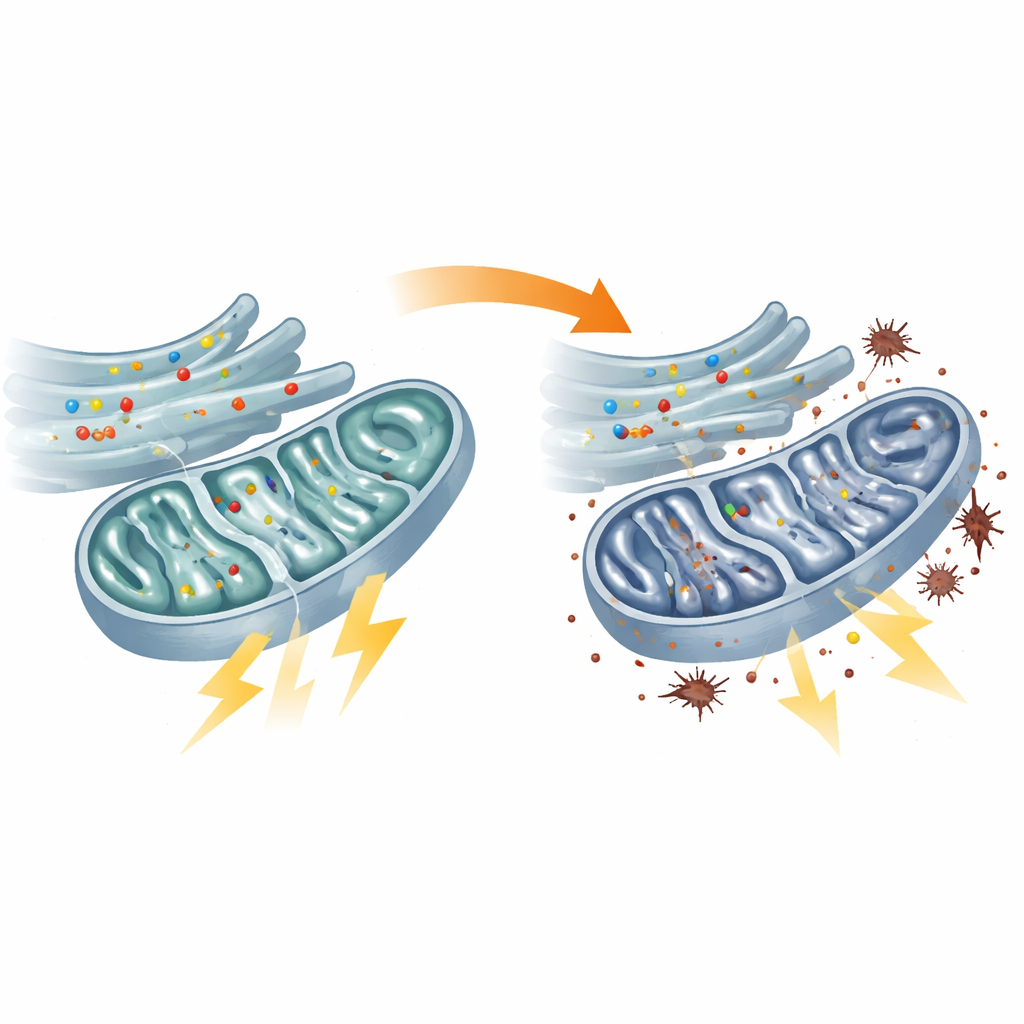
Mitochondrien verhungert und Backup-Systeme überlastet
Direkte Tests des Energiestoffwechsels bestätigten, dass die Mitochondrien in den Fibroblasten der Patientinnen und Patienten unterperformten. Der zentrale Treibhaus-Prozess, der Zitronensäurezyklus (TCA-Zyklus), lief langsamer, mehrere seiner Schlüsselenzyme waren weniger aktiv, und die Reaktionskette, die normalerweise Treibstoff in ATP — die Energieeinheit der Zelle — umwandelt, war gedämpft. Infolgedessen fielen die ATP-Werte innerhalb der Mitochondrien. Die Zellen versuchten auszugleichen, indem sie die Glykolyse hochfuhren, einen weniger effizienten, zuckerverbrennenden Weg außerhalb der Mitochondrien. Dieser Umschwung hatte seinen Preis: Elektronen «leckten» aus der gestressten mitochondrialen Maschinerie und lösten erhöhte Lipidperoxidation aus, eine Form oxidativer Schädigung der Zellmembranen. Ähnliche Defekte zeigten sich in Zebrafischen mit reduziertem flvcr1a, was den direkten Zusammenhang zwischen mitochondrialem Versagen und dem Tiermodell der sensorischen Neuropathie herstellt.
Hinweise auf zukünftige Therapien durch Stärkung der Zellenergie
Ermutigend ist, dass sich einige dieser Defekte im Labor lindern ließen. Als das Team den Calcium-Einstrom in Mitochondrien künstlich erhöhte, indem es das Kanalprotein MCU in Patientenzellen überexprimierte, erholte sich die Energieproduktion und die Anzeichen oxidativer Schäden nahmen ab. Die Versorgung der Zellen mit einem Häm-Vorläufer, 5-Aminolevulinsäure (ALA), verbesserte ebenfalls die Aktivität des TCA-Zyklus, die Funktion der Atmungskette und die ATP-Werte, obwohl längere ALA-Exposition in früheren Studien schädlich war. Zusätzlich normalisierte extra Cholin die Membranfluidität und half, Lipidschäden zu verringern, brachte aber nur mäßige kurzfristige Verbesserungen der Energieproduktion. Diese Rescue-Experimente legen nahe, dass kein einzelner Weg allein verantwortlich ist; vielmehr treibt ein Netzwerk aus gestörtem Cholin-, Häm- und Calciumstoffwechsel die Mitochondrien in eine chronische Unterleistung.
Warum diese Erkenntnisse für Patientinnen und Patienten wichtig sind
Indem die Folgen von FLVCR1-Mutationen von Molekülen über Zellen bis zum ganzen Organismus verfolgt werden, legt diese Arbeit nahe, dass Energieversagen in Mitochondrien eine treibende Kraft hinter dieser Form der Schmerzverlust-Neuropathie und den damit verbundenen Sehproblemen ist. Sensorische Nerven und Photorezeptoren sind außergewöhnlich energieintensiv, weil sie lange Axone aufrechterhalten oder lichtempfindliche Strukturen kontinuierlich erneuern, und sind daher besonders verletzlich, wenn die mitochondriale Leistung nachlässt. Das Zebrafisch-Modell und die patientenabgeleiteten Zellen bieten jetzt praxisnahe Testplattformen für Therapien, die den mitochondrialen Stoffwechsel stärken. Während Behandlungen wie Cholin-Supplementierung, kontrolliertes Häm-Boosting oder Wirkstoffe, die die mitochondriale Calciumaufnahme verbessern, sorgfältige Prüfung in Tiermodellen und klinischen Studien erfordern, ist die zentrale Botschaft klar: Die Wiederherstellung der Energieversorgung empfindlicher Neurone könnte eines Tages helfen, Menschen zu schützen, die ohne das wichtigste Warnsignal der Natur — Schmerz — geboren wurden.
Zitation: Bertino, F., Zanin Venturini, D.I., Grasso, E. et al. Mitochondrial energetic failure underlies FLVCR1-related sensory neuropathy. Commun Biol 9, 429 (2026). https://doi.org/10.1038/s42003-026-09691-y
Schlüsselwörter: sensorische Neuropathie, mitochondriale Dysfunktion, FLVCR1, Schmerzunempfindlichkeit, Nerven-Energiestoffwechsel