Clear Sky Science · de
Abnorme Eisen‑Homöostase vermittelt Schädigung von Cochlea‑Haarsinneszellen und Hörverlust bei Gprasp2‑defizienten Mäusen
Warum das für das Hören im Alltag wichtig ist
Hörverlust wird oft als einfache „Abnutzung“ verstanden, doch bei vielen Menschen liegt die Ursache in versteckten Gendefekten. Diese Studie zeigt, wie ein wenig untersuchtes Gen, GPRASP2, die empfindlichen schallwahrnehmenden Zellen des Innenohrs vor eisengetriebener Schädigung schützt. Indem sie exakt darlegt, was schief läuft, wenn dieses Gen fehlt, eröffnet die Arbeit Möglichkeiten für präzisere Diagnosen und künftige Therapien bei erblichen Hörstörungen und möglicherweise auch bei damit zusammenhängenden Stimmungserkrankungen.
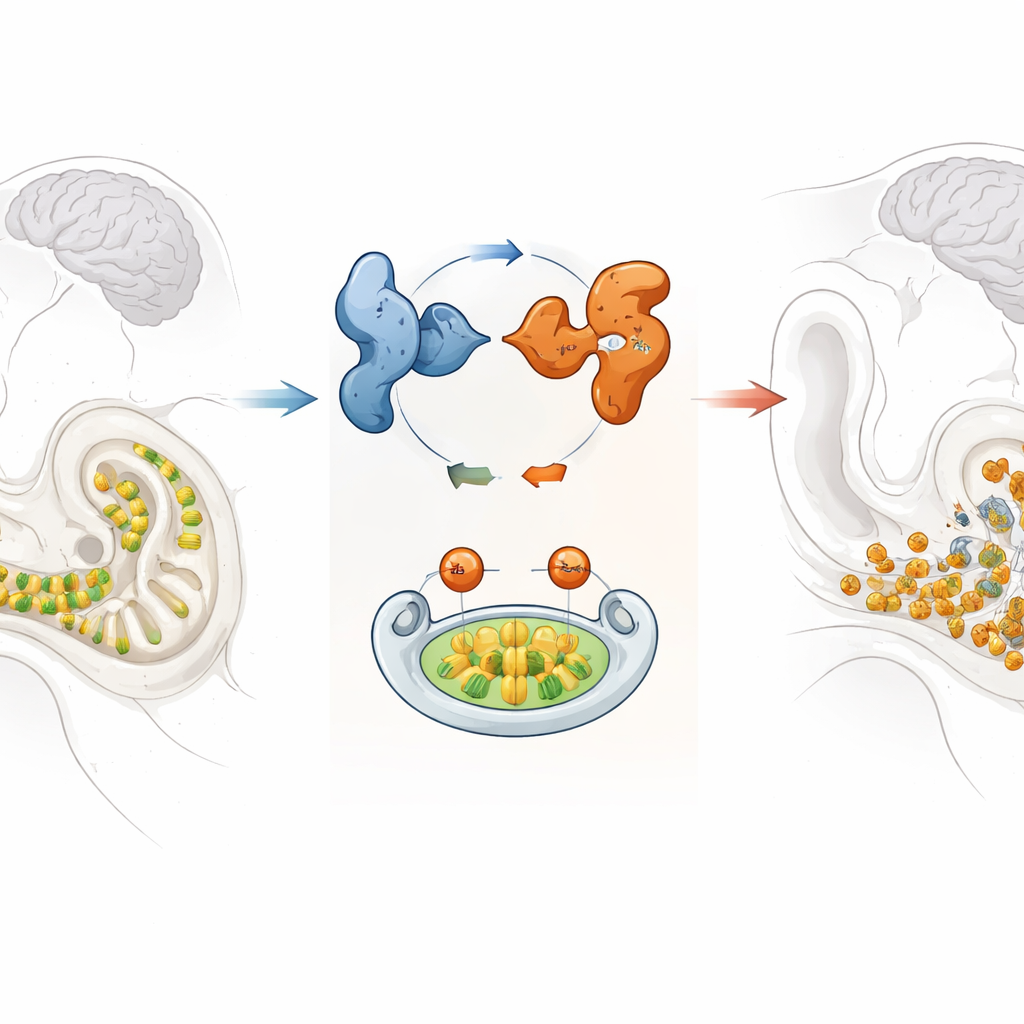
Ein fragiler Schallsensor im Innenohr
Im gewundenen Cochlea des Innenohrs sitzen Reihen von Haarsinneszellen, die winzige Vibrationen in elektrische Signale umwandeln, die das Gehirn versteht. Gehen diese Haarsinneszellen verloren, wachsen sie nicht nach, daher ist ihre Erhaltung für lebenslanges Hören entscheidend. Die Forscher konzentrierten sich auf GPRASP2, ein Gen, das zuvor in einer Familie mit X‑chromosomal vererbtem syndromalem Hörverlust verändert gefunden wurde – eine Erkrankung, die vorwiegend Männer betrifft und mit zusätzlichen Symptomen einhergehen kann. Obwohl bekannt war, dass GPRASP2 im Gehirn und im Innenohr aktiv ist, war seine genaue Rolle für das Hören unklar.
Was passiert, wenn das Gen fehlt
Um die menschliche Situation nachzuahmen, nutzte das Team CRISPR‑Geneditierung, um Mäuse zu erzeugen, denen eine funktionsfähige Kopie des Gprasp2‑Gens fehlt. Diese Tiere zeigten bei empfindlichen elektrischen Messungen des Hörnervs deutlichen Hörverlust über ein breites Frequenzspektrum. Ihre Reaktionen auf plötzliche laute Geräusche waren abgeschwächt, während Gleichgewicht und Motorik größtenteils normal blieben – ein Hinweis auf ein spezifisches Problem des Hörens statt einer allgemeinen Bewegungsstörung. Interessanterweise zeigten die Mäuse in mehreren Standardtests auch depressionähnliches Verhalten, was darauf hindeutet, dass dieses Gen Hören und Stimmung durch seine Wirkung im Ohr und im Gehirn verknüpfen kann.
Schäden im Inneren der Cochlea
Bei der Untersuchung der Innenohren von Gprasp2‑defizienten Mäusen fanden die Wissenschaftler, dass viele äußere Haarsinneszellen fehlten oder fehlplatziert waren und die verbleibenden Zellen oft verzerrte Bündel winziger Fortsätze aufwiesen, die normalerweise wie Stimmgabeln wirken. Sie beobachteten auch Veränderungen in der Stria vascularis, einem hochaktiven Gewebe, das die spezielle Flüssigkeits‑ und elektrische Umgebung für das Hören aufrechterhält. Auf mikroskopischer Ebene waren Marker für Zelltod und oxidativen Stress in den äußeren Haarsinneszellen und den benachbarten Nervenzellen erhöht, was anzeigt, dass diese Strukturen stark von reaktiven Molekülen angegriffen wurden und eher zum Absterben neigen.
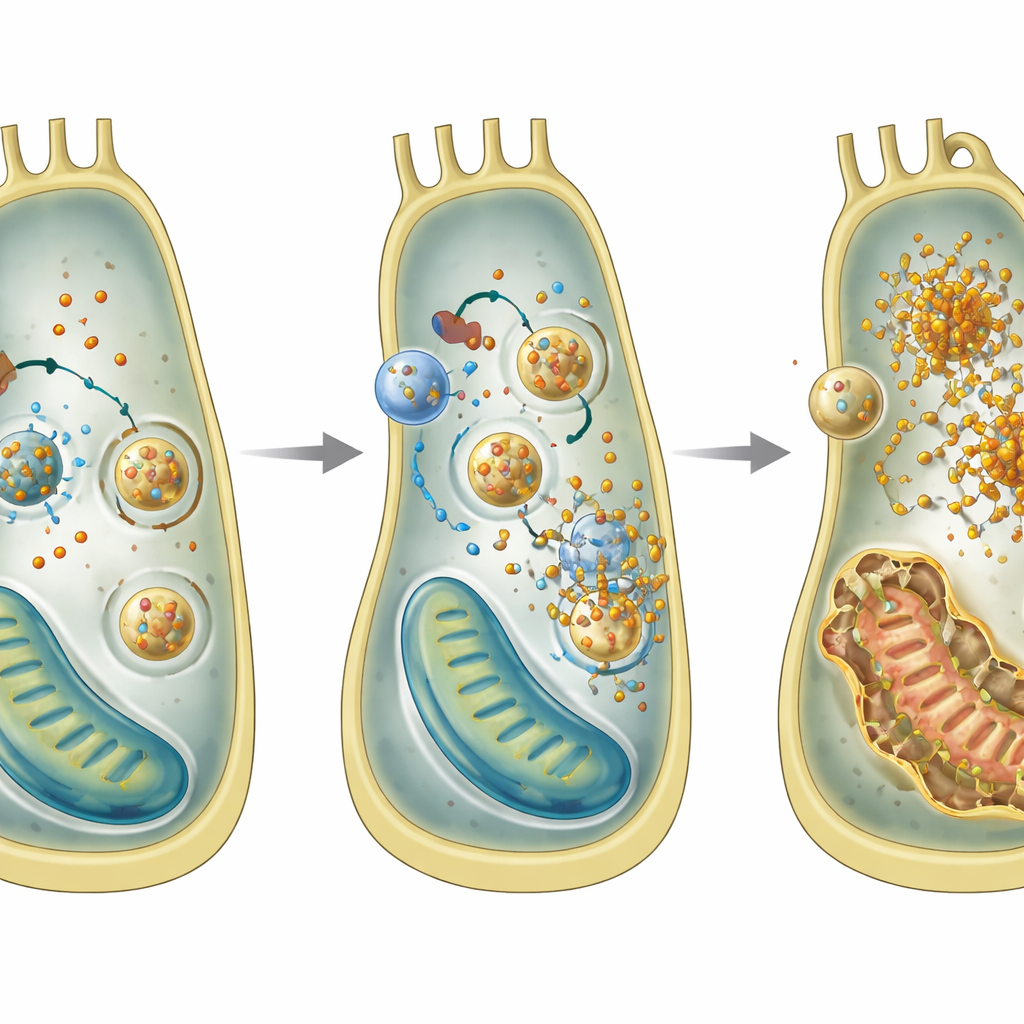
Eisenüberladung und außer Kontrolle geratene Wiederverwertung
Tiefergehend verwendete das Team eine Zelllinie des Innenohrs, um zu untersuchen, was in einzelnen Zellen ohne GPRASP2 schiefgeht. Sie entdeckten ein Muster, das typisch ist für eine Form des Zelltods namens Ferroptose, die durch Eisen und Lipidperoxidation angetrieben wird. Zellen ohne Gprasp2 häuften mehr zweiwertiges Eisen an, produzierten mehr reaktive Sauerstoffspezies, wiesen niedrigere Glutathion‑Spiegel auf und zeigten geschädigte Mitochondrien. Gen‑ und Proteinanalysen deuteten auf eine Zunahme der Ferritinophagie hin – eines Prozesses, bei dem Eisen‑Speicherpartikel an Recycling‑Kompartimente geliefert und dort abgebaut werden, wodurch zusätzliches freies Eisen freigesetzt wird. Die Blockade dieses Recycling‑Schritts reduzierte die Eisenakkumulation und stützt die Idee, dass übermäßige Ferritinophagie zentral für die Schädigung ist.
Ein wichtiger Partnerprotein, das Eisen im Zaum hält
Die Forscher fragten dann, wie GPRASP2 diese eisenregulierenden Mechanismen steuert. Durch Kartierung von Proteinen, die mit GPRASP2 interagieren, identifizierten sie NCAM1, ein Zelladhäsionsmolekül, das vor allem für seine Rollen bei Lernen, Gedächtnis und Stimmung bekannt ist. Sie zeigten, dass GPRASP2 physisch an NCAM1 bindet und dass der Verlust von GPRASP2 zu verringerten NCAM1‑Spiegeln in Haarsinneszellen und in kultivierten auditorischen Zellen führt. Niedrigeres NCAM1 war mit erhöhter Ferritinophagie und Eisenüberladung verknüpft. Die Wiederherstellung von NCAM1 in Gprasp2‑defizienten Zellen senkte die Eisenwerte und dämpfte zentrale Ferritinophagie‑Regulatoren, obwohl die allgemeine Autophagie aktiv blieb. Das legt nahe, dass GPRASP2 einen spezifischen eisenrecyclingbezogenen Weg über NCAM1 feinreguliert, statt das gesamte Recycling‑System ein- oder auszuschalten.
Was das für das Hören und darüber hinaus bedeutet
Einfach gesagt zeigt diese Arbeit, dass GPRASP2 wie ein Sicherheitsmanager für Eisen in Cochlea‑Haarsinneszellen wirkt. Wenn GPRASP2 vorhanden ist, arbeitet es mit NCAM1 zusammen, um zu verhindern, dass zu viel gespeichertes Eisen wieder in die Zelle freigesetzt wird, und hält so oxidativen Schaden in Schach. Ist das Gen gestört, gerät die Eisenhandhabung aus dem Gleichgewicht, die Zellen „rosten“ von innen und entscheidende Haarsinneszellen sterben ab, was zum Hörverlust führt. Da GPRASP2 und NCAM1 auch im Gehirn funktionieren, kann derselbe Weg die bei einigen Patienten beobachteten Stimmungsschwankungen erklären. Das Verständnis dieses eisenbasierten Versagensmechanismus bietet ein klares Ziel für zukünftige Medikamente oder Gentherapien, die darauf abzielen, das Hören bei Menschen mit GPRASP2‑bezogenen oder ähnlichen genetischen Störungen zu bewahren.
Zitation: Lu, Y., Sheng, F., Yao, J. et al. Abnormal iron homeostasis mediates cochlear hair cell impairment and hearing loss in Gprasp2-deficient mice. Commun Biol 9, 425 (2026). https://doi.org/10.1038/s42003-026-09679-8
Schlüsselwörter: erbliche Schwerhörigkeit, Cochlea‑Haarsinneszellen, Eisen‑Homöostase, Ferroptose, GPRASP2