Clear Sky Science · de
Ein praxisorientierter Leitfaden zu gezielten Single-Cell-RNA-Sequenzierungstechnologien
Warum der Blick auf einzelne Zellen wichtig ist
Jede Zelle in Ihrem Körper trägt dieselbe DNA, doch verschiedene Zellen verhalten sich sehr unterschiedlich. Das erreichen sie, indem sie bestimmte Gene ein- oder ausschalten und RNA‑Moleküle auf subtile Weise verändern. Moderne Single-Cell-RNA-Sequenzierung kann bei Tausenden von Zellen gleichzeitig ablesen, welche RNAs vorhanden sind, verpasst aber derzeit große Teile der Botschaft. Dieser Übersichtsartikel erklärt, wo heutige Methoden Informationen verlieren und wie neue „gezielte“ Verfahren entwickelt werden, um für Forschung, Diagnose und Therapieplanung gezielt die wichtigsten Bereiche von RNA‑Molekülen ins Visier zu nehmen.
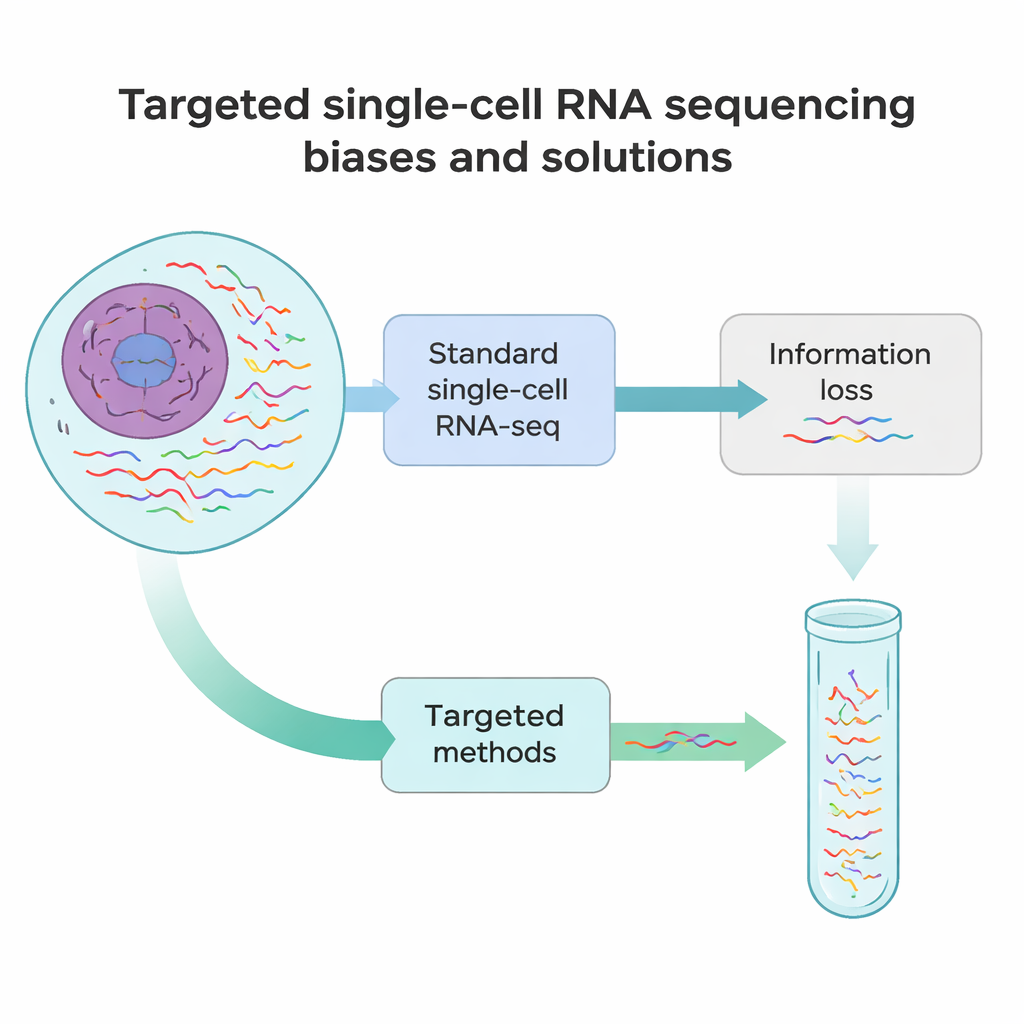
Worin heutige Methoden zurückbleiben
Standardmäßige Single-Cell-RNA-Sequenzierung ähnelt eher dem Anfertigen eines kurzen Schnappschusses jeder Nachricht in einer Zelle als dem Abspielen eines vollständigen Films. In den meisten Experimenten werden nur etwa 10–40 % aller RNAs in einer Zelle nachgewiesen, und meist wird nur deren Anfang oder Ende gelesen. Das bedeutet, dass viele seltene, aber wichtige RNAs – etwa Marker, die die Identität einer Zelle bestimmen, oder Genvarianten mit krankheitsverursachenden Mutationen – leicht übersehen werden. Hinzu kommen mehrere technische Schritte, vom Zerlegen von Gewebe in Einzelzellen bis zum Umkopieren von RNA in DNA und der Amplifikation, die systematische Verzerrungen einführen. Manche RNAs werden früh abgeschnitten, andere überrepräsentiert, und wieder andere verschwinden vollständig aus den Daten.
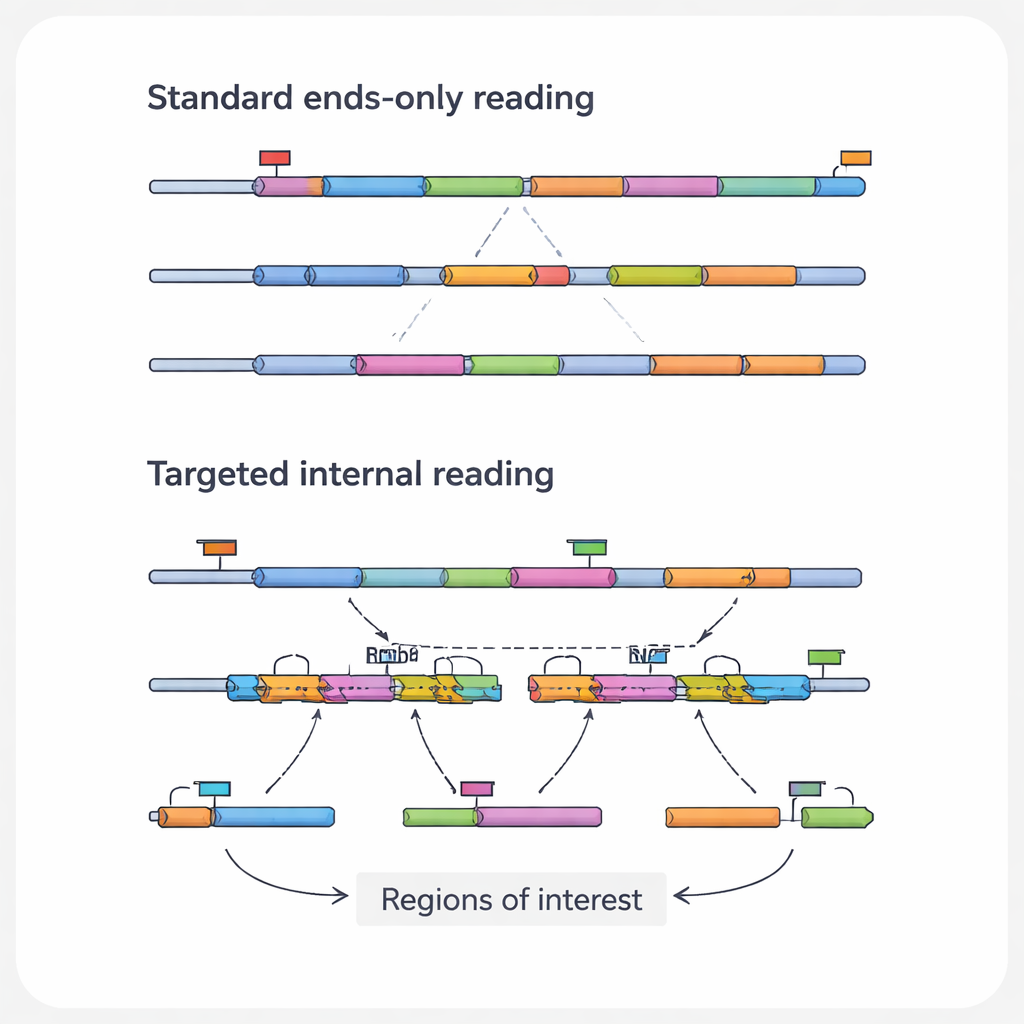
Warum interne RNA-Details wichtig sind
Die medizinisch relevantesten Informationen in einem RNA‑Molekül liegen oft in seinen inneren Bereichen, nicht an den Enden, die Standardmethoden sehen. Diese inneren Abschnitte können Punktmutationen enthalten, die Krebs antreiben, Fusionsstellen, an denen zwei Gene untypisch verbunden wurden, oder Spleißverknüpfungen, die verschiedene Proteinvarianten aus demselben Gen erzeugen. Sie können auch die Spuren von Genomeditierungswerkzeugen wie CRISPR dokumentieren. Die Autoren bezeichnen diese spezifischen Merkmale als „Interessensregionen“ und die RNAs, die sie tragen, als „Transkripte von Interesse“. Da gängige Hochdurchsatzplattformen vorwiegend die Spitzen der RNAs lesen, übersehen sie routinemäßig diese entscheidenden Details, insbesondere bei langen oder gering präsentierten Transkripten.
Neue Wege, den Scheinwerfer zu richten
Um diese blinden Flecken zu überwinden, haben Forschende eine Familie gezielter Single-Cell-RNA-Sequenzierungsansätze entwickelt. Anstatt zu versuchen, jede RNA gleich zu lesen, reichern diese Methoden gezielt ausgewählte Transkripte oder Regionen an. Manche Strategien gestalten die Fängerkügelchen so um, dass sie an interne RNA‑Sequenzen binden statt nur an das Poly(A)-Ende und ziehen ausgewählte Botschaften bereits im ersten Schritt in die Bibliothek. Andere fügen kundenspezifische Primer hinzu, die das Kopieren an einer internen Stelle starten, oder zusätzliche PCR‑Schritte, die eine Kurzliste von Genen aus einer bestehenden Bibliothek gezielt amplifizieren. Wieder andere nutzen DNA‑Sonden, die an Ziel‑RNAs oder deren Kopien hybridisieren und diese dann oft mit einfachen chemischen Markern herausfischen. Jede Kategorie tauscht Sensitivität, Zellzahl, Anzahl der Ziele und Kosten gegeneinander aus, aber alle verfolgen dasselbe Ziel: aus denselben oder weniger Sequenzierungsreads aussagekräftigere Details zurückzugewinnen.
Anwendungen von Viren bis zu Tumoren
Diese gezielten Methoden verändern bereits mehrere Bereiche der Biologie und Medizin. Bei Infektionen können sie endlich virale oder bakterielle RNAs erfassen, die keine Poly(A)-Schwänze besitzen, wie sie Standardprotokolle erwarten, und zeigen, welche Wirtszellen sie bewohnen und wie sie die Wirtsgenaktivität verändern. In der Krebsforschung kann gezielte Single-Cell-Sequenzierung präzise bestimmen, welche Zelltypen spezifische Mutationen oder Fusionsgene tragen und diese mit veränderten Genprogrammen verknüpfen, was hilft zu erklären, warum manche Zellen therapieresistent werden. Andere Methoden konzentrieren sich auf alternatives Spleißen und legen offen, welche Zelltypen welche Isoformen nutzen, oder auf seltene Zellpopulationen und subtile Marker, die sonst unter der Nachweisgrenze blieben. In gepoolten CRISPR‑Screens erlaubt eine verbesserte Erfassung der Guide‑RNAs, jede genetische Störung mit ihrer exakten zellulären Antwort zu verbinden.
Die richtige Methode wählen und Ausblick
Da es inzwischen eine breite Auswahl an gezielten Ansätzen gibt, schlagen die Autoren einen Entscheidungsbaum vor, der Forschenden bei der Wahl einer Methode hilft. Zentrale Fragen sind, ob eine vollständige Transkriptomprofilierung nötig ist, wie viele Gene oder Regionen anvisiert werden müssen, wie weit diese Regionen von den RNA‑Enden entfernt liegen und wie viele Zellen verarbeitet werden können. Mit Blick auf die Zukunft argumentieren sie, dass die größten Fortschritte von der Verbesserung der allerersten Erfassungsschritte, der Ausweitung cleverer Sonden‑basierter Strategien und der Kombination von Targeting mit aufkommenden Long‑Read‑ und Direkt‑RNA‑Sequenzierungsplattformen zu erwarten sind. Bis es praktikabel wird, jede RNA in jeder Zelle von Anfang bis Ende zu lesen, bleibt die gezielte Single-Cell-RNA-Sequenzierung unerlässlich, um die Teile der zellulären Botschaft sichtbar zu machen, die für Biologie und Krankheit am wichtigsten sind.
Zitation: Moro, G., Brunner, E. & Basler, K. A practical guide to targeted single-cell RNA sequencing technologies. Commun Biol 9, 250 (2026). https://doi.org/10.1038/s42003-026-09675-y
Schlüsselwörter: Single-Cell-RNA-Sequenzierung, gezielte Sequenzierung, Transkriptomik, Krebs‑Mutationen, räumliche Transkriptomik