Clear Sky Science · de
DANST ermöglicht die Zelltypprozession in der räumlichen Transkriptomik mithilfe tiefen domänenadversarialer neuronaler Netze
Zellen in ihrem Umfeld sehen
Humane Gewebe sind dicht bevölkerte Städte aus vielen verschiedenen Zelltypen, von denen jeder eine eigene Funktion übernimmt. Neue „räumliche Transkriptomik“-Technologien können messen, welche Gene über einen Gewebeschnitt hinweg aktiv sind, aber jede Messung vermischt oft Signale mehrerer benachbarter Zellen. Diese Arbeit stellt DANST vor, eine intelligente rechnerische Methode, die diese Mischungen auseinanderdröselt. Indem sie uns sagt, welche Zelltypen vorhanden sind und wo sie sich in Organen und Tumoren befinden, hilft sie Forschenden, besser zu verstehen, wie Gewebe aufgebaut sind, wie Krankheiten sich ausbreiten und wo Behandlungen am besten ansetzen könnten. 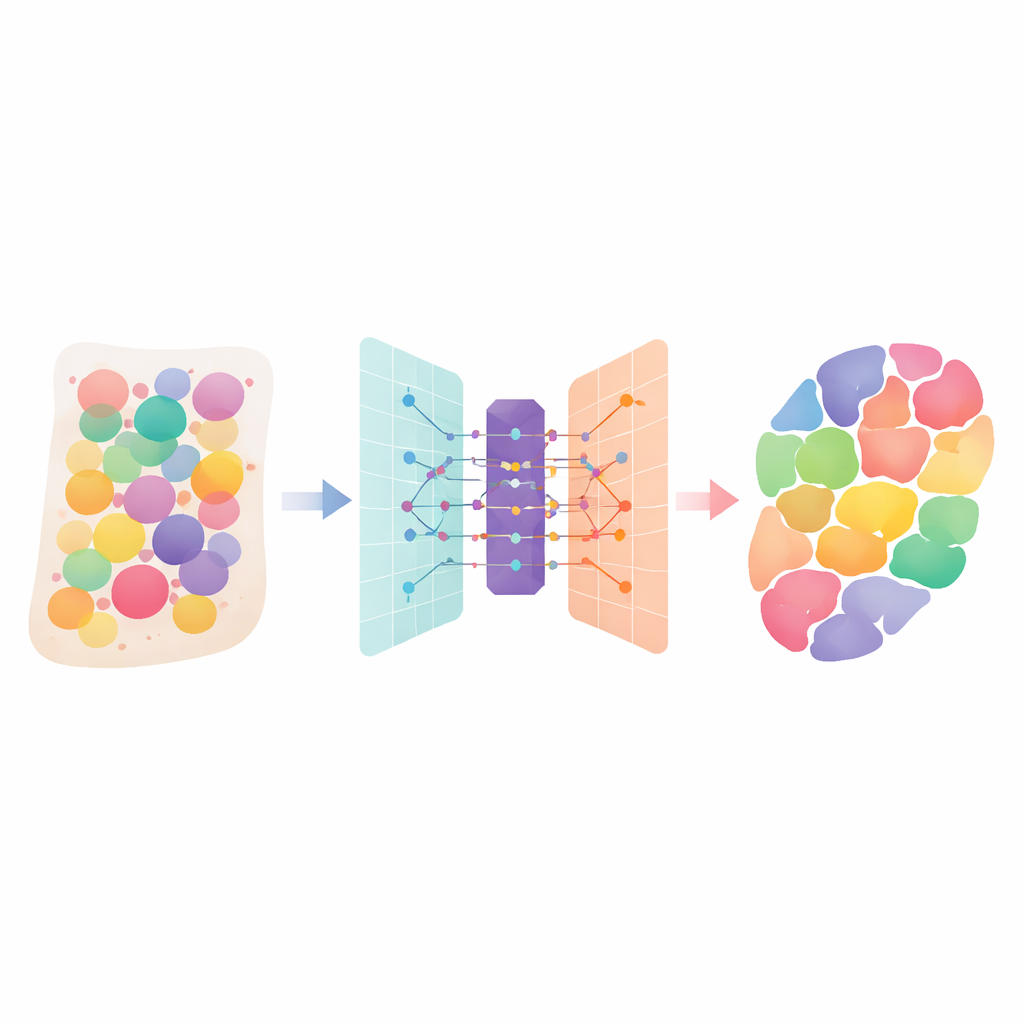
Die Herausforderung, dichte Zellansammlungen zu entwirren
Moderne Genlese-Methoden können entweder sehr präzise einzelne Zellen betrachten oder das vollständige Layout eines Gewebes erfassen, selten aber beides gleichzeitig. Gängige räumliche Technologien zeichnen Genaktivität in relativ großen „Spots“ auf, die mehrere Zellen umfassen können. Das Ergebnis ist vergleichbar mit einem Chor, bei dem man hört, was gesungen wird, aber nicht, wer welche Töne singt. Um das zu interpretieren, benötigen Forschende Dekonvolutionsmethoden, die schätzen, welchen Anteil jeder Zelltyp an jedem Spot hat. Viele bestehende Ansätze nutzen Single-Cell-Daten als Referenz, doch sie haben Probleme, weil die beiden Datentypen in unterschiedlichen Experimenten erhoben werden und sich in Qualität, Rauschen und Auflösung nicht perfekt entsprechen.
Eine Brücke zwischen Datenwelten bauen
DANST geht dieses Missverhältnis an, indem es eine Brücke zwischen Single-Cell- und räumlichen Daten schafft. Zunächst nutzt es die detaillierten Single-Cell-Profile, um viele künstliche gemischte Spots mit bekannten Zelltypanteilen zu simulieren. Gleichzeitig gruppiert es die realen räumlichen Spots entsprechend ihrer Lage im Gewebe und ihrer Genmuster und verwendet die Abstände zu diesen Gruppen, um jedem simulierten Spot eine „Pseudo“-Position zuzuweisen. Dieser Schritt erzeugt eine verknüpfte Karte, in der künstliche und reale Spots einen gemeinsamen räumlichen Bezugsrahmen teilen, wodurch die Methode lernen kann, wie gemischte Signale in bestimmten Nachbarschaften des Gewebes aussehen sollten. 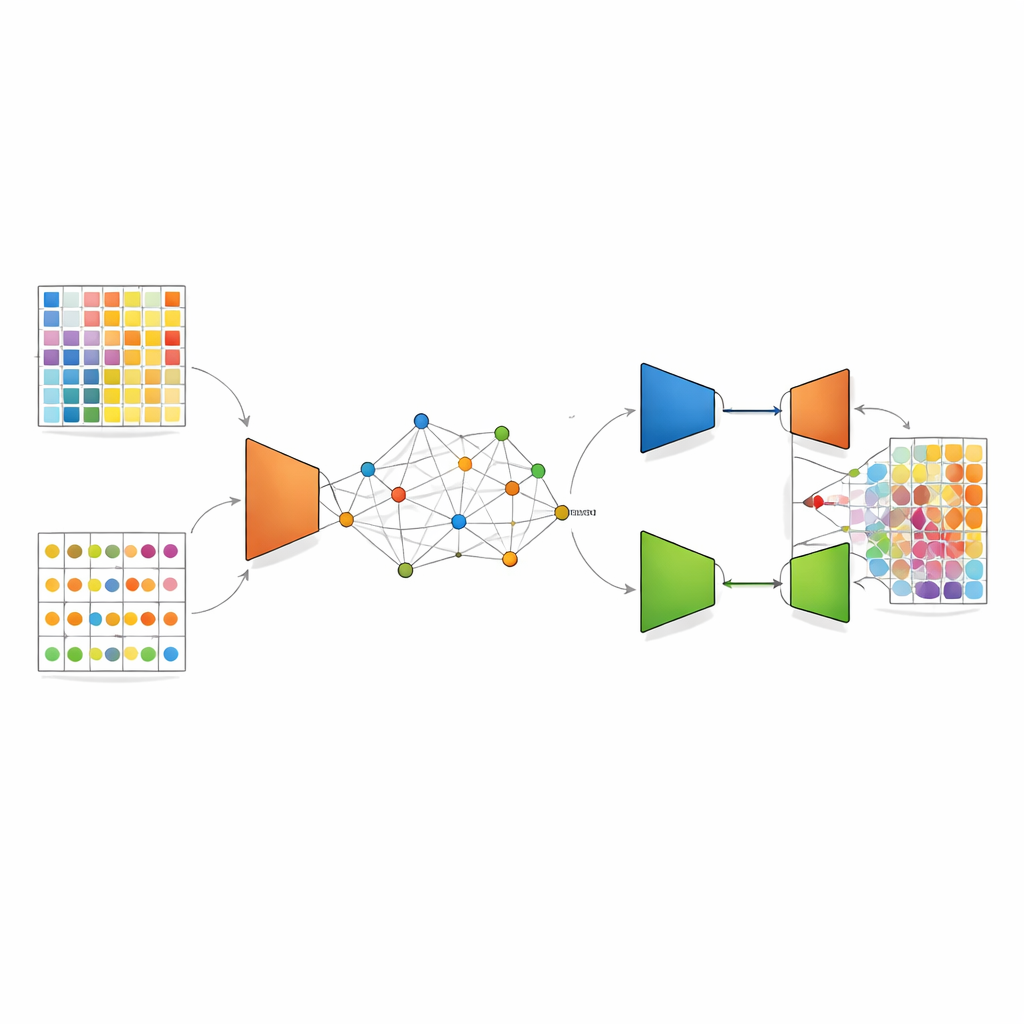
Signale bereinigen und Domänen ausrichten
Sobald diese gemeinsame Karte vorliegt, wendet DANST eine Form des Deep Learning namens variationaler Autoencoder an. Dieses Netzwerk komprimiert Genmuster aus sowohl realen als auch simulierten Spots zu einer verfeinerten internen Repräsentation und versucht dann, sie zu rekonstruieren, wodurch Daten effektiv entrauscht und wichtige Merkmale hervorgehoben werden. Darauf setzt die Arbeit eine adversariale Komponente: Ein zweites Netzwerk versucht zu erkennen, ob ein verfeinertes Muster aus realen räumlichen Daten oder aus simulierten Daten stammt, während der Merkmalsextraktor lernt, es zu täuschen. Dieses „Tauziehen“ treibt das Modell dahin, Merkmale zu finden, die für beide Datenquellen gut funktionieren, sodass aus simulierten Spots mit bekannten Zelltypanteilen gewonnenes Wissen zuverlässig auf reale Gewebespots übertragen werden kann, deren Zusammensetzung unbekannt ist.
Tests an Herzen, Gehirnen und Tumoren
Das Team testete DANST an künstlichen Benchmarks sowie an realen biologischen Proben von Mäusen und Menschen. Im Vergleich zu mehreren führenden Methoden stellte DANST die Zelltypanteile in synthetischen Datensätzen genauer wieder her und behauptete seinen Vorteil über sehr unterschiedliche Gewebe und Plattformen hinweg. In einem Mausgehirn-Datensatz rekonstruierte es klar die geschichtete Organisation des Kortex und stimmte mit fachkundig definierten anatomischen Regionen überein. In einem separaten Mausgehirnschnitt erfasste es feine Muster in Bereichen wie dem Hippocampus. Besonders bemerkenswert: In humanem Brustkrebsgewebe zeichnete DANST nach, wie verschiedene Immunzellen, Stütz- und hormonempfindliche luminale Zellen innerhalb und um Tumorregionen angeordnet sind. Diese Karten entsprachen bekannter Biologie und deuteten auf klinisch relevante Merkmale hin, etwa Hormonabhängigkeit und potenziell ungünstigere Prognosen dort, wo bestimmte Immunzellen rar waren.
Was das für Biologie und Medizin bedeutet
Für Nicht-Fachleute lässt sich DANST als ein leistungsfähiger Übersetzer betrachten, der verschwommene, überlappende Signale in ein klares Bild verwandelt, welche Zellen wo in einem Gewebe leben. Indem Zelltypen räumlich zuverlässig getrennt werden, bietet es Forschenden eine schärfere Sicht darauf, wie gesunde Organe organisiert sind und wie Krankheiten diese Organisation verändern. In der Krebsforschung kann dies aufdecken, wie Tumorzellen und Immunzellen in spezifischen Regionen interagieren, was gezielte Therapien leiten und die Vorhersage von Patientenverläufen verbessern kann. Mit dem zunehmenden Angebot an räumlichen und Single-Cell-Datensätzen sind Werkzeuge wie DANST darauf vorbereitet, unverzichtbar zu werden, um die zellulären Nachbarschaften zu entschlüsseln, die Gesundheit und Krankheit zugrunde liegen.
Zitation: Zhang, X., Wu, Z., Wang, T. et al. DANST enables cell-type deconvolution in spatial transcriptomics using deep domain adversarial neural networks. Commun Biol 9, 388 (2026). https://doi.org/10.1038/s42003-026-09659-y
Schlüsselwörter: räumliche Transkriptomik, Dekonvolution von Zelltypen, Deep Learning, Tumormikromilieu, Single-Cell-RNA-Sequenzierung