Clear Sky Science · de
Molekulare QTL sind in einem Rinder-Langread-Kohortenbestand für Strukturvarianten angereichert
Warum Rinder-DNA uns etwas über komplexe Merkmale lehren kann
Bauern, Tierärzte und Genetiker möchten alle verstehen, warum manche Tiere schneller wachsen, resistenter gegen Krankheiten sind oder mehr Milch geben als andere. Ein großer Teil der Antwort liegt in der DNA, aber unsere üblichen Werkzeuge betrachten meist winzige Veränderungen einzelner "Buchstaben" des Genoms. Diese Studie zeigt, dass deutlich größere DNA-Veränderungen – Strukturvarianten – still und heimlich die Genfunktion bei Rindern formen und dass neue Langread-Sequenzierungstechnologien uns endlich ermöglichen, ihr volles Ausmaß zu erkennen.
Das Genom mit schärferer Linse betrachten
Die meisten genetischen Studien stützen sich auf kurze DNA-Abschnitte, die günstig und präzise sind, aber in repetitiven oder komplexen Regionen des Genoms an ihre Grenzen stoßen. Die Autor:innen verwendeten eine neuere Methode, Langread-Sequenzierung, an 120 Bullen einer milchbezogenen Rinderrasse. Diese langen Reads überbrücken deutlich größere DNA-Strecken und erleichtern so das Auffinden großer Insertions-, Deletions- und Umordnungsereignisse, die als Strukturvarianten bekannt sind. Das Team verglich diese Langreads mit vorhandenen Short-Read-Daten derselben Tiere und stellte fest, dass Langreads insgesamt mehr Varianten aufdeckten und die Abdeckung schwieriger Regionen wie der X- und Y-Chromosomen deutlich verbesserten.
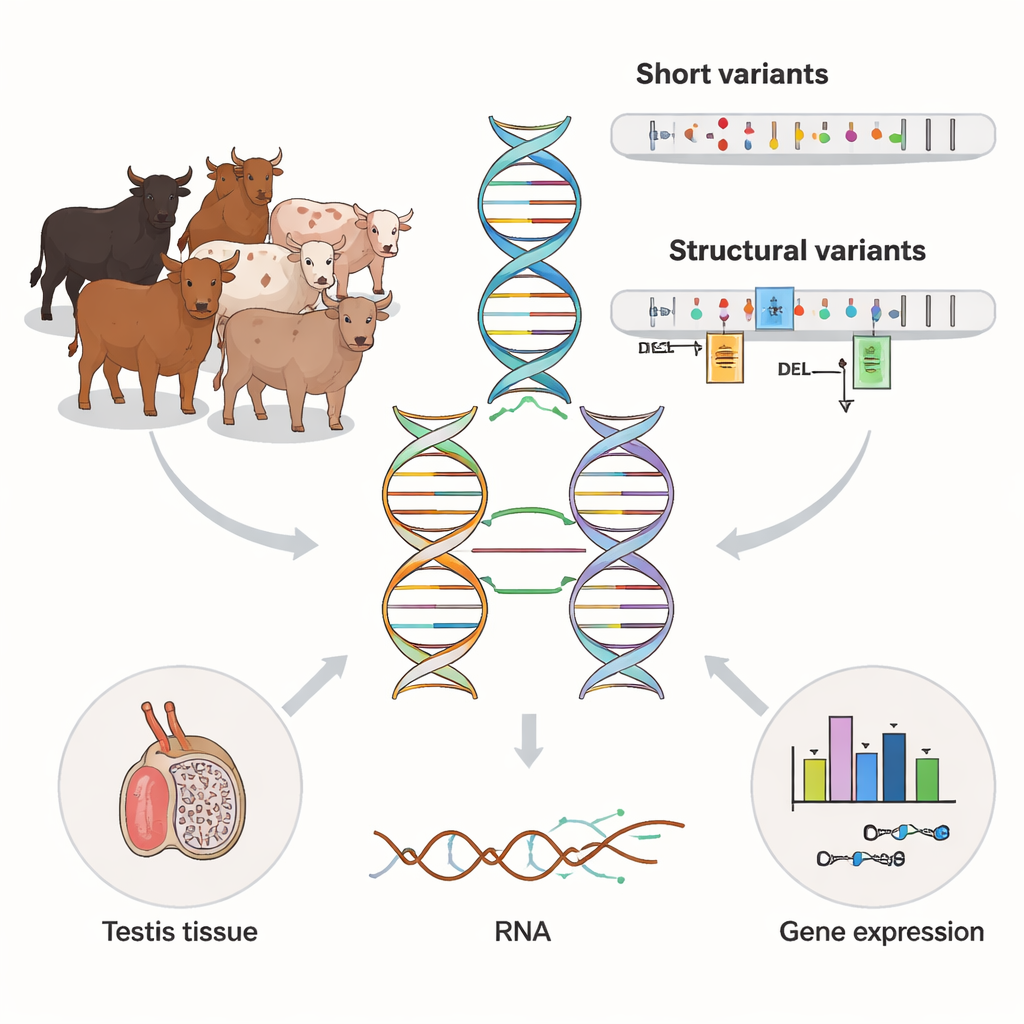
Aufdeckung tausender verborgener DNA-Umordnungen
Mit den Langread-Daten katalogisierten die Forschenden etwa 24 Millionen kleine DNA-Veränderungen und über 79.000 Strukturvarianten über die Bullen hinweg. Viele dieser größeren Veränderungen standen in Verbindung mit repetitiven DNA-Elementen, die sich im Genom kopieren und einsetzen. Etwa eine von zehn Strukturvarianten trat nur in einem oder zwei Tieren auf und zeigte damit ein reiches Reservoir seltener Variation. Im Vergleich zu einem früheren Rinder‑Pangenom, das aus hochwertigen Assemblies erstellt wurde, ergänzte der neue Datensatz zehntausende zusätzliche Strukturvarianten, insbesondere Insertionsereignisse und komplexe Duplikationen, die mit älteren Methoden schwer zu erfassen sind. Das deutet darauf hin, dass Langread-Studien weiterhin zuvor unsichtbare Schichten genetischer Vielfalt im Nutztierbereich aufdecken.
Verknüpfung von DNA-Veränderungen mit Genaktivität
Um zu untersuchen, wie diese DNA-Unterschiede die Biologie tatsächlich beeinflussen, richtete das Team den Blick auf ein Gewebe, das für männliche Fruchtbarkeit wichtig ist: den Hoden. Für 117 der Bullen lagen tiefe RNA-Sequenzierungsdaten vor, die zeigen, welche Gene angeschaltet sind und wie ihre Transkripte gespleißt werden. Durch statistisches Verknüpfen von genetischen Varianten in der Nähe jedes Gens mit dessen Aktivität identifizierten sie über 27.000 "molekulare QTLs" – genomische Stellen, die entweder die Menge eines exprimierten Gens oder die Art des RNA‑Spleißens verändern. Strukturvarianten traten als wichtige Einflussgrößen hervor: Sie waren mehr als doppelt so häufig unter den stärksten Expressionssignalen und mehr als fünfmal so häufig unter den stärksten Spleißsignalen wie per Zufall zu erwarten. In vielen Fällen war die einflussreichste Variante eine große Insertion, Deletion oder Duplikation in einem Promoter, Enhancer, Exon oder Spleißstellenbereich und nicht ein Einzelbuchstabenwechsel.
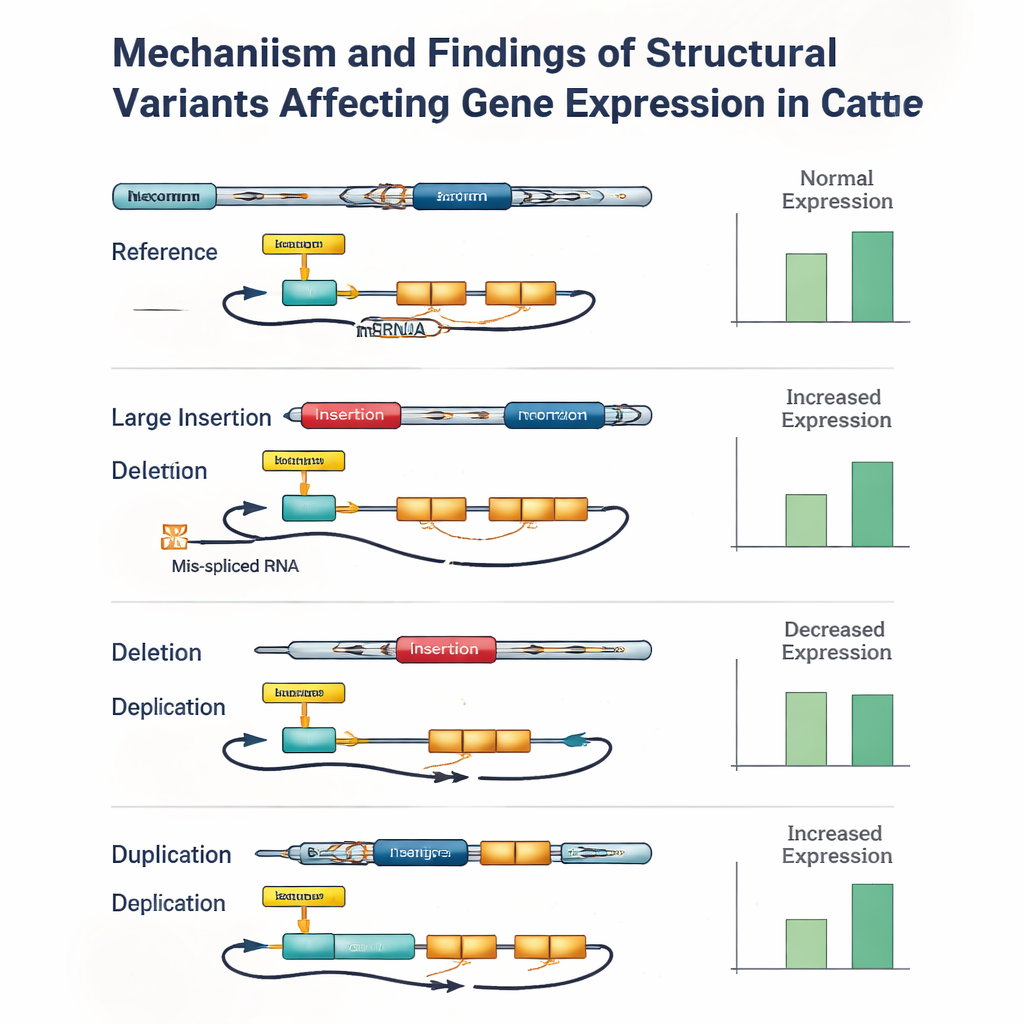
Wenn Genotypisierungsfehler wichtige Signale verbergen
Die Studie legte jedoch auch die Grenzen aktueller Werkzeuge offen. Selbst bei hochwertigen Langreads war die genaue Zuordnung von Strukturvarianten-Genotypen zu einzelnen Tieren herausfordernd, besonders bei großen Insertionsereignissen und langen Duplikationen. Kleine Fehler – gelegentlich nur bei einem oder zwei Bullen – konnten dazu führen, dass eine Strukturvariante statistisch etwas schwächer erschien als eine nahegelegene kleine Variante, die perfekt mit ihr gekoppelt war. Als die Autor:innen einige der stärksten Signale manuell überprüften, fanden sie wiederholt Fälle, in denen eine Strukturvariante innerhalb eines Gens oder einer wichtigen regulatorischen Region die plausibelste Ursache des Effekts war, aber Genotypisierungsfehler oder fehlende Daten eine gekoppelte kleine Variante an die Spitze der Rangliste setzten.
Was das für Zucht und darüber hinaus bedeutet
Für Nicht‑Fachleute lautet die Quintessenz: "große" DNA‑Veränderungen sind sehr bedeutsam. Diese Langread‑Bestandsaufnahme bei Rindern zeigt, dass Strukturvarianten stark unter den genetischen Stellen angereichert sind, die steuern, wie Gene ein- und ausgeschnitten werden, insbesondere im Reproduktionsgewebe. Gleichzeitig warnt die Studie, dass heutige Analyseverfahren viele dieser Varianten noch übersehen oder falsch kennzeichnen, vor allem bei moderater Sequenzierungstiefe. Mit sinkenden Kosten und steigender Genauigkeit der Langread‑Sequenzierung sowie verbesserter Software werden Züchter und Forschende in der Lage sein, ökonomisch wichtige Merkmale – wie Fruchtbarkeit, Krankheitsresistenz und Milchleistung – bis auf konkrete Strukturvarianten zurückzuverfolgen. Dieselben Prinzipien gelten für die menschliche Gesundheit und Pflanzenzüchtung: Um komplexe Merkmale vollständig zu verstehen, müssen wir über Einzelbuchstabenänderungen hinausblicken und die größeren Umordnungen berücksichtigen, die Genome neu formen.
Zitation: Mapel, X.M., Leonard, A.S. & Pausch, H. Molecular QTL are enriched for structural variants in a cattle long-read cohort. Commun Biol 9, 290 (2026). https://doi.org/10.1038/s42003-026-09596-w
Schlüsselwörter: Strukturvarianten, Langread-Sequenzierung, Rindergenomik, Genexpression, molekulare QTL