Clear Sky Science · de
Zellbasierte und isoformselektive Assays für G-Protein-gekoppelte Rezeptorkinasen zur umfassenden Evaluierung von Inhibitoren
Warum das Herunterregeln zellulärer „Lautstärkeregler“ wichtig ist
Viele unserer Medikamente wirken, indem sie die Aktivität von Zelloberflächenrezeptoren, die Hormone, Neurotransmitter und Arzneistoffe wahrnehmen, hoch- oder runterdrehen. Diese Rezeptoren müssen anschließend sorgfältig wieder abgeschaltet werden, damit Zellen nicht überstimuliert bleiben – ein Prozess, der teilweise von Enzymen namens GRKs gesteuert wird. Sind GRKs überaktiv, wie bei Herzinsuffizienz und einigen Krebsarten, gerät die Signalübertragung aus dem Gleichgewicht. Diese Studie entwickelt praktikable, zellbasierte Tests, um zu messen, wie gut experimentelle Moleküle spezifische GRKs blockieren können, und hilft so Forschern, intelligentere Wirkstoffe zu entwerfen, die diese wichtigen zellulären Lautstärkeregler feinjustieren.
Tore an der Zelloberfläche
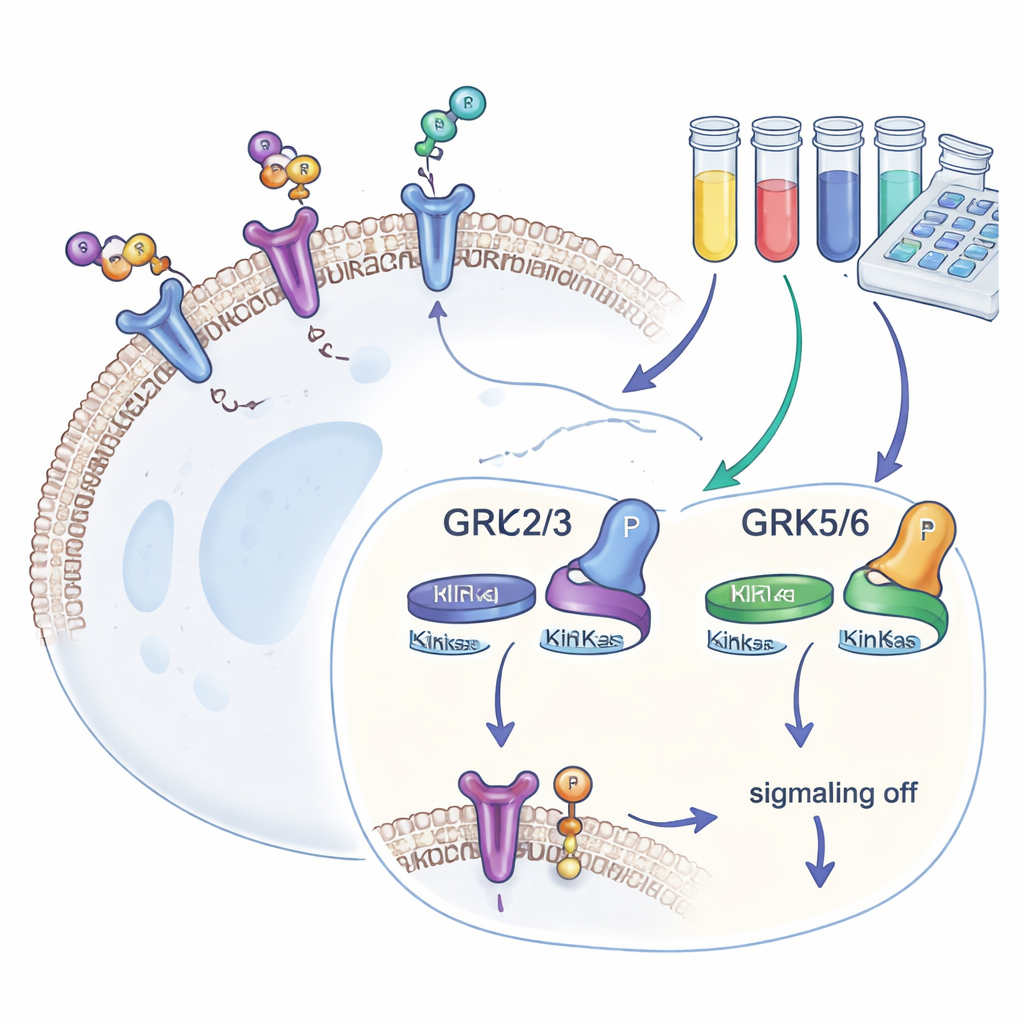
Unsere Zellen tragen Hunderte von Typen G-Protein-gekoppelter Rezeptoren (GPCRs), die äußere Signale erkennen und in innere Antworten umsetzen. Nachdem ein GPCR ausgelöst wurde, heften GRKs kleine Phosphat-„Marken“ an sein Ende. Diese Marken ziehen ein weiteres Protein, beta-Arrestin, an, das die weitere Signalgebung stoppt und den Rezeptor häufig in die Zelle hineinzieht. Vier GRK-Varianten – GRK2, GRK3, GRK5 und GRK6 – kommen in vielen Geweben vor. Weil sie beeinflussen, wie stark GPCRs reagieren, und weil ihre Mengen bei Krankheiten wie Herzinsuffizienz, Krebs und Suchterkrankungen verändert sind, sind Wirkstoffentwickler daran interessiert, GRK-Blocker zu finden, die sowohl wirksam als auch selektiv sind.
Ein sauberes Testfeld innerhalb von Zellen aufbauen
Die meisten früheren GRK-Studien beruhten auf Computermodellen oder Reagenzglaschemie, die zeigen, wie stark ein Inhibitor binden kann, aber nicht, wie er sich im dichten Inneren einer lebenden Zelle verhält. Um diese Lücke zu schließen, konstruierten die Autoren menschliche HEK293-Zellen, denen alle vier häufigen GRKs fehlen, und führten dann jeweils nur eine GRK-Isoform wieder ein. Jede Zelllinie trug außerdem einen gut untersuchten Rezeptor, den Beta-2-adrenergen Rezeptor, der so markiert war, dass seine Phosphorylierung an einer spezifischen C-terminalen Stelle (genannt T360/S364) mittels eines empfindlichen, Antikörper-basierten Assays abgelesen werden konnte. Da diese Stelle nur von GRKs modifiziert wird, dient die vorhandene Phosphatmenge als direkte, quantitative Messgröße für die Aktivität jeder GRK-Isoform in lebenden Zellen.
Die Guten, die Schwachen und die Unspezifischen trennen
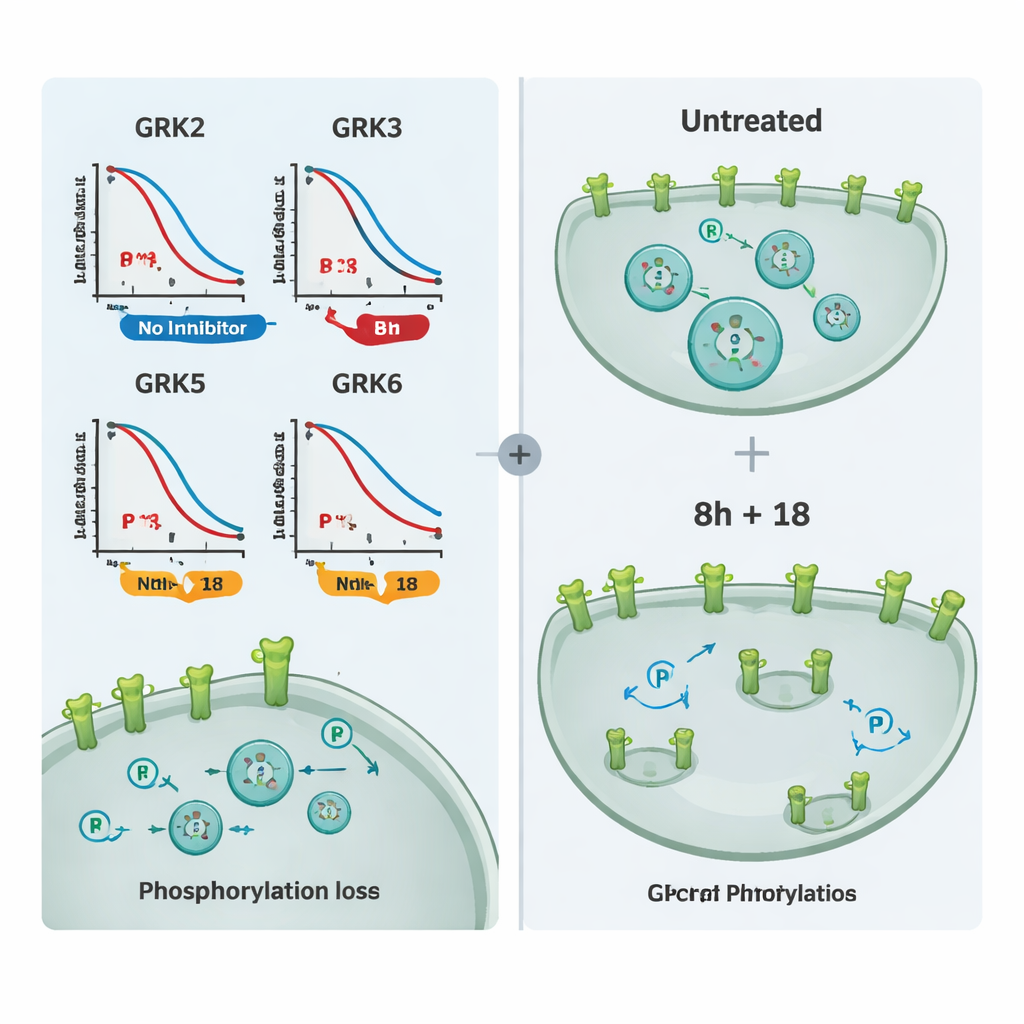
Mithilfe dieses Werkzeugkastens testete das Team eine Reihe kommerziell verfügbarer GRK-Blocker. Zuerst gruppierten sie Verbindungen, die überwiegend GRK2 und GRK3 anvisieren, und eine andere Gruppe, die auf GRK5 und GRK6 abzielt. Durch den Vergleich, wie stark jedes Molekül die Rezeptorphosphorylierung in Zellen reduzierte, die nur einen GRK-Subtyp exprimierten, konnten sie die Selektivität in der Praxis abbilden. Eine Verbindung mit der Bezeichnung 8h erwies sich als der potenteste Hemmer von GRK2/3, während Verbindung 18 sich als selektiver Inhibitor von GRK5/6 hervortat. Einige weit verbreitete Moleküle zeigten in den getesteten Konzentrationen wenig Wirkung, vermutlich weil sie nur schlecht in Zellen eindringen, und ein sehr stark wirkender kovalenter Inhibitor beeinträchtigte die Zellgesundheit, sodass er für Bildgebungs‑Experimente ungeeignet war.
Von chemischen Fingerabdrücken zum Rezeptorverhalten
Um zu zeigen, dass diese Inhibitoren nicht nur einen Testrezeptor betreffen, sondern die GPCR-Biologie allgemein beeinflussen, untersuchten die Autoren mehrere medizinisch relevante Rezeptoren, darunter den mu-Opioidrezeptor und den Vasopressin-V2-Rezeptor. Sie maßen sowohl Phosphorylierung als auch Rezeptorinternalisierung mittels Mikroskopie. Die alleinige Behandlung mit 8h oder 18 verringerte bei vielen Zielen teilweise die Phosphorylierung und das Einwärtsbewegen der Rezeptoren, aber die Kombination von 8h und 18 verhinderte diese Veränderungen nahezu vollständig und hielt die Rezeptoren an der Zelloberfläche. Zusätzliche Experimente zur Rekrutierung von beta-Arrestin bestätigten, dass dieselben Verbindungen die Signalgebung an anderen Rezeptoren regulieren können, die von überlappenden GRK-Sets kontrolliert werden.
Was das für künftige Arzneimittel bedeutet
Für Nichtfachleute ist die Kernbotschaft, dass die Studie ein verlässliches Set zellbasierter Tests liefert – und zwei besonders nützliche Werkzeugverbindungen, 8h und 18 –, mit denen Forscher in lebenden Zellen genau erkennen können, wie unterschiedliche GRK-Isoformen heruntergeregelt werden. Statt aus vereinfachten Reagenzglasdaten zu spekulieren, können Wissenschaftler Kandidateninhibitoren nun nebeneinander vergleichen und entscheiden, ob sie hauptsächlich GRK2/3, GRK5/6 oder alle vier zugleich beeinflussen. Diese Klarheit sollte die Entwicklung von Medikamenten beschleunigen, die die GPCR-Signalübertragung präziser modulieren, mit potenziellen Vorteilen für die Behandlung von Herzkrankheiten, Krebs, Schmerzsyndromen und anderen Erkrankungen, bei denen das Gleichgewicht der Signalgebung gestört ist.
Zitation: Blum, N.K., Kiefer, M.C., Decker, A. et al. Cell-based and isoform-selective G protein-coupled receptor kinase assays for comprehensive inhibitor evaluation. Commun Biol 9, 287 (2026). https://doi.org/10.1038/s42003-026-09568-0
Schlüsselwörter: GPCR-Signalübertragung, GRK-Inhibitoren, beta-adrenerger Rezeptor, zellbasierter Assay, Arzneimittelentdeckung