Clear Sky Science · de
haCCA: multi-modulare Integration von spot-basierten spatialen Transkriptomen und Metabolomen
Warum es wichtig ist, Moleküle am Ort zu kartieren
Unser Körper besteht aus zahllosen kleinen Nachbarschaften von Zellen, jede mit ihrer eigenen Mischung aktiver Gene und chemischer Substanzen. Bis vor Kurzem mussten Wissenschaftler diese Moleküle untersuchen, indem sie Gewebe zu einer einheitlichen Paste zerkleinerten und damit jeglichen räumlichen Kontext verloren. Diese Arbeit stellt eine neue rechnerische Methode vor, haCCA, die zwei leistungsfähige Bildgebungstechniken zusammenführt, sodass Forschende in situ sehen können, wie Gene und kleine Moleküle in realen Geweben und Tumoren angeordnet sind. Solche Karten können verborgene Krankheitsmuster aufdecken und präzisere Behandlungsansätze nahelegen.
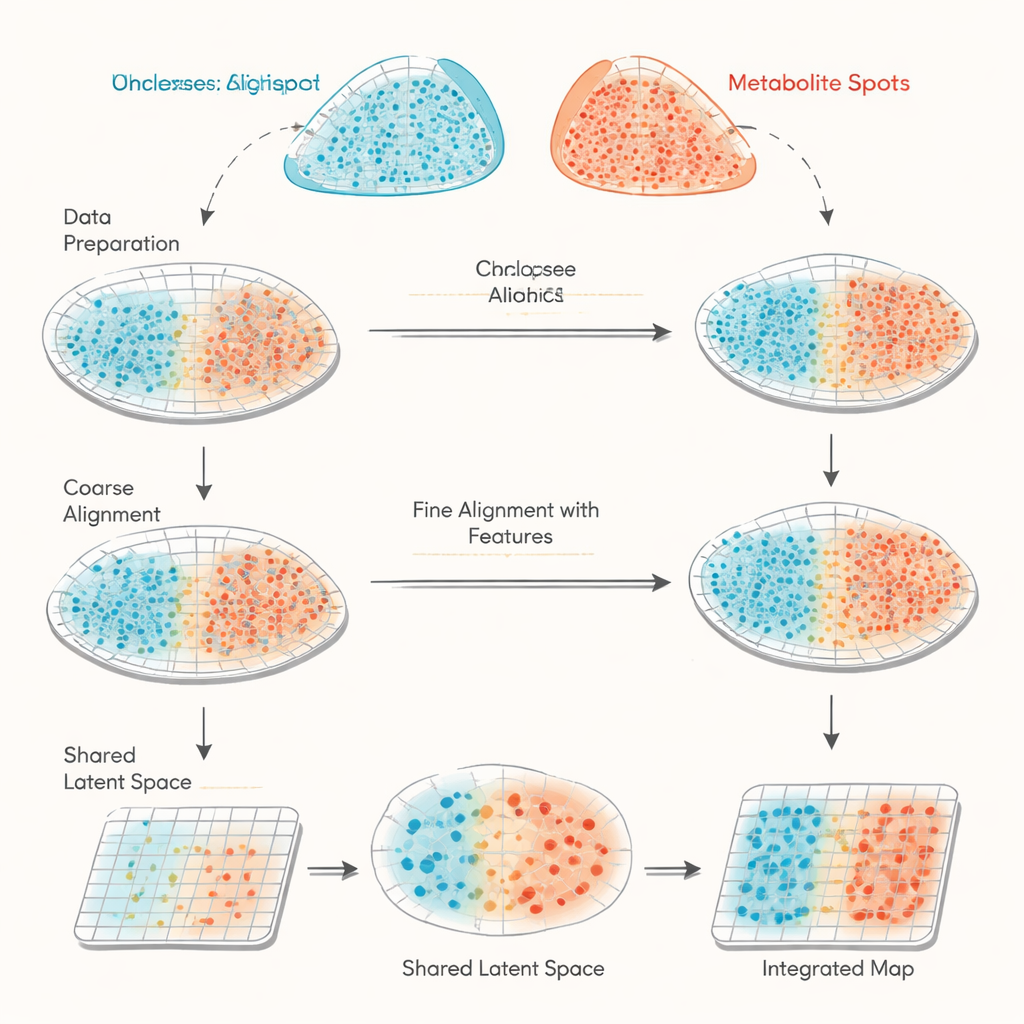
Zwei verschiedene Ansichten desselben Gewebes
Die Studie konzentriert sich auf die Kombination von Daten aus zwei räumlichen Methoden, die in der Biologie zunehmend eingesetzt werden. Spatial Transcriptomics zeichnet auf, welche Gene in tausenden kleiner Spots über einen Gewebeschnitt aktiviert sind. MALDI-Massenspektrometrie-Imaging erfasst die Mengen vieler kleiner Moleküle, etwa Metaboliten und Lipide, an ähnlich dichten Rasterpunkten. Das Problem ist, dass diese beiden Instrumente nicht exakt dieselben Positionen oder denselben Satz von Merkmalen messen, sodass ihre Daten wie zwei fehljustierte Karten mit unterschiedlichen Legenden sind. Bestehende Ansätze versuchen meist, die Formen der Gewebeschnitte allein anhand ihrer Koordinaten abzugleichen, was ungenau sein kann und keine gute Möglichkeit bietet, die Qualität der Ausrichtung zu überprüfen.
Ein klügerer Weg, molekulare Karten auszurichten
haCCA (kurz für hierarchical anchor-guided canonical correlation analysis) geht dieses Problem an, indem es Geometrie und Biologie kombiniert. Zuerst führt es eine zweistufige „morphologische Ausrichtung“ der Spot-Raster beider Technologien durch. Menschliche Expertinnen und Experten wählen einige übereinstimmende Landmarken in den Gewebebildern, um grobe Verschiebungen und Rotationen zu korrigieren; ein automatischer Schritt verfeinert anschließend Ausreißer in der Nähe eingerissener Ränder oder fehlender Teile. Danach sucht die Methode nach „Anker“-Paaren von Spots, die räumlich nahe beieinander liegen und sich in lokal homogenen Regionen befinden, sodass sie wahrscheinlich denselben Gewebebereich repräsentieren. Aus diesen Anker-Spots berechnet haCCA, welche Gene und Metaboliten typischerweise gemeinsam variieren, und verdichtet diese Informationen zu einer gemeinsamen nieder-dimensionalen Darstellung, die ihre stärksten gemeinsamen Muster einfängt.
Korrelationen in ein einheitliches Gewebebild übertragen
Mit sowohl räumlichen Koordinaten als auch der gemeinsamen molekularen Darstellung löst haCCA ein Optimierungsproblem, um zu entscheiden, wie wahrscheinlich es ist, dass jeder Gen-Spot mit jedem Metaboliten-Spot übereinstimmt. Dieser Schritt ist so gestaltet, dass Spots räumlich nahe beieinander bleiben, aber auch in ihrem kombinierten Gen–Metabolit-Profil ähnlich sind. Das Endergebnis ist ein „Transportplan“, der jeden Punkt eines Datensatzes mit seinem besten Partner im anderen verknüpft und so eine integrierte multimodale Karte erzeugt. An sorgfältig konstruierten Testdaten — bei denen die wahren Beziehungen bekannt sind — zeigen die Autorinnen und Autoren, dass jede Phase des Workflows (grobe Ausrichtung, verfeinerte Ausrichtung und merkmalsbewusstes Matching) drei unabhängige Genauigkeitsmaße schrittweise verbessert. Verglichen mit anderen Werkzeugen, die hauptsächlich auf Geometrie basieren, erreicht haCCA konsequent höhere Ausrichtungsgenauigkeit und eine treuere Übertragung von Regionsbezeichnungen.
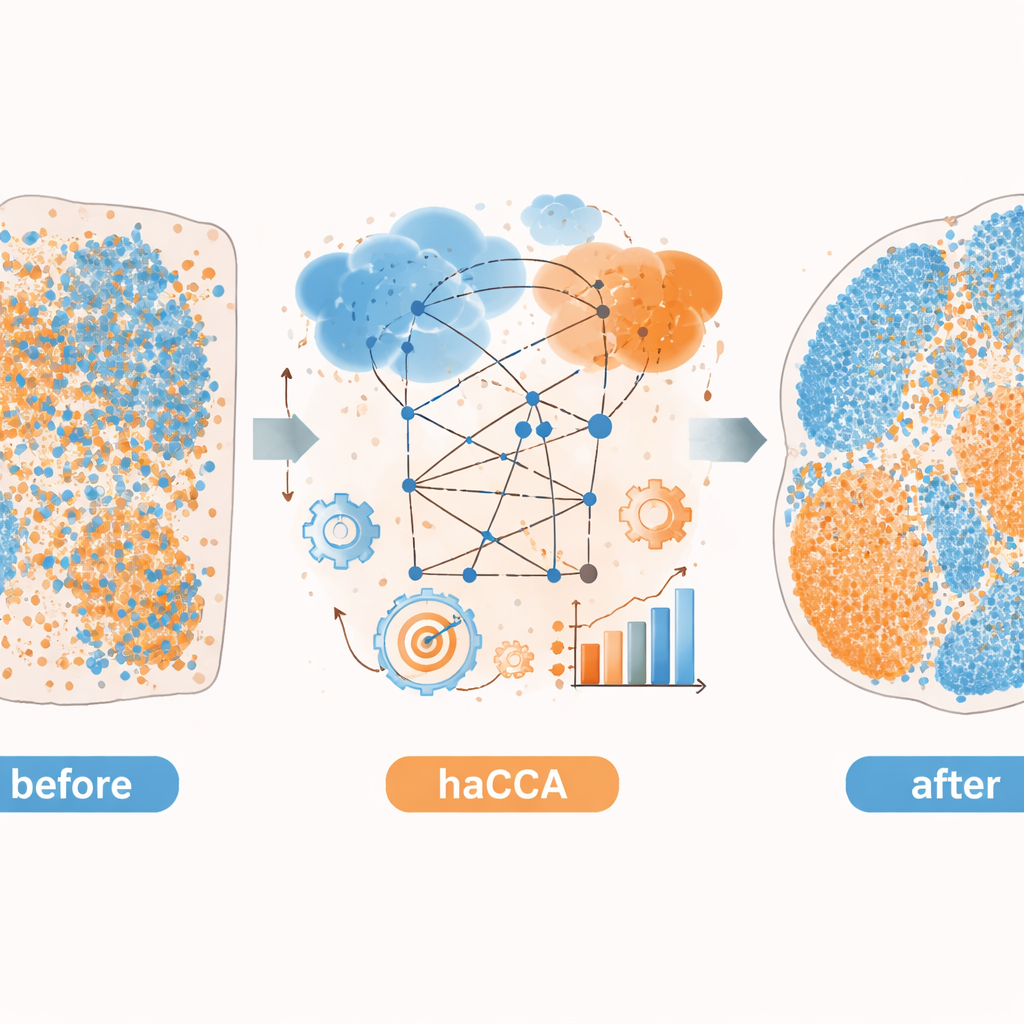
Verborgene Biologie in Gehirn- und Lebertumoren sichtbar machen
Die Autorinnen und Autoren wenden haCCA anschließend auf echte Mausgewebe aus Gehirn und Lebertumoren an. Für das Gehirn integrieren sie kommerzielle Spatial-Transcriptomics-Daten mit Metabolitenbildern aus denselben oder benachbarten Schnitten. Die Methode erhält bekannte metabolische Territorien und rekonstruiert erwartete Überlappungen, etwa die Ko-Lokalisierung von Dopamin mit dem Gen, das sein Schlüsselenzym kodiert. Durch gemeinsames Clustern von Genen und Metaboliten finden sie, dass die kombinierten Daten subtilere Gewebeunterregionen unterscheiden als jede Modalität für sich allein. In einem präklinischen Modell des intrahepatischen Cholangiokarzinoms, einer Form von Leberkrebs, nutzen sie haCCA, um Tumoren zu vergleichen, die neutrophile extrazelluläre Fallen — netzartige Strukturen, die von Immunzellen freigesetzt werden — bilden können oder nicht. Die integrierten Karten zeigen, dass bei Anwesenheit dieser Fallen ein Gen namens Scd1 und dessen zugehörige Fettsäuren in malignen Regionen angereichert sind, was auf eine Verschiebung hin zu verändertem Fettstoffwechsel im Tumor hindeutet.
Was das für zukünftige Forschung bedeutet
Anschaulich ist haCCA wie das Übereinanderlegen von Luftaufnahmen, die mit verschiedenen Kameras gemacht wurden — die eine sensibel für Gebäudekonturen, die andere für Wärmesignaturen — um ein schärferes Bild dessen zu erhalten, was in jedem Häuserblock vor sich geht. Indem es genau zusammenführt, wo Gene aktiv sind und wo sich wichtige Metaboliten anreichern, hilft dieser Workflow Forschenden, beide Seiten zellulären Verhaltens zugleich zu profilieren: die Instruktionen und die daraus resultierende Chemie. Der Ansatz verbessert frühere Ausrichtungsmethoden, ist als zugängliches Python-Tool verpackt und lässt sich auf andere räumliche Technologien ausdehnen. Mit zunehmender Verbreitung solcher integrierten Karten könnten wir ein tieferes Verständnis dafür gewinnen, wie Tumoren und andere Gewebe ihren Stoffwechsel organisieren, auf Behandlungen reagieren und sich im Laufe der Zeit verändern.
Zitation: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Schlüsselwörter: räumliche Multi-Omics, Transkriptomik, Metabolomik, Tumorstoffwechsel, Datenintegration