Clear Sky Science · de
Untersuchung der Wechselwirkungen zwischen dem Immunitätsprotein Colicin E9 von Escherichia coli und der DNA-Gyrase von Pseudomonas aeruginosa: fortgeschrittener computergestützter Ansatz zur Entwicklung neuartiger antimikrobieller Strategien
Bakterielle Waffen in neue Medikamente verwandeln
Während sich die Antibiotikaresistenz ausbreitet, gehen Ärzten die Mittel aus, um gefährliche Infektionen zu bekämpfen. Manche der hartnäckigsten Erreger, etwa Pseudomonas aeruginosa, sind gegen viele Wirkstoffe unempfindlich. Diese Studie sucht in den Bakterien selbst nach neuen Ideen und untersucht, ob das eingebaute Schutzprotein eines Mikroorganismus dazu umfunktioniert werden kann, ein lebenswichtiges Enzym in einem anderen Mikroorganismus zu deaktivieren. Mithilfe fortgeschrittener Computersimulationen zeigen die Forschenden, wie ein kleines „Immunitäts“-Protein fest an ein zentrales bakterielles Enzym binden kann, und deuten damit einen potenziellen neuen Weg für künftige Antimikrobika an.
Ein winziger Schutzschild gegen ein tödliches Toxin
Bestimmte Stämme von Escherichia coli produzieren potente Protein-Toxine, sogenannte Colicine, die benachbarte Bakterien töten können. Um sich nicht selbst zu vergiften, stellen diese Bakterien passende Immunitätsproteine her. Eines dieser Schutzproteine, bekannt als das Colicin E9-Immunitätsprotein (häufig Im9 genannt), bindet an die katalytische Domäne des Toxins und verhindert so Schäden an der DNA des Wirts. Da diese Partnerschaft so spezifisch und stark ist, vermuteten Wissenschaftler lange, dass ihr detailliertes Verständnis neue Wege zur Kontrolle schädlicher Bakterien offenbaren könnte. In dieser Arbeit fragen die Autorinnen und Autoren, ob Im9 möglicherweise auch an die DNA-Gyrase, ein essentielles Enzym in Pseudomonas aeruginosa, binden kann, das die Wicklung und Replikation seiner DNA steuert.
Ein verletzliches Enzym in einem schwer zu behandelnden Keim anvisieren
Pseudomonas aeruginosa ist ein bedeutender Krankenhauskeim, der in rauen Umgebungen überleben kann und gegen viele Wirkstoffe resistent ist. Die DNA-Gyrase ist eines seiner wichtigsten Enzyme und sorgt dafür, dass die langen DNA-Stränge des Bakteriums korrekt verdrillt bleiben, damit sie repliziert werden können. Da die Hemmung dieses Enzyms das Bakterienwachstum stoppen kann, ist es bereits Ziel einiger Antibiotika. Die Autorinnen und Autoren nutzten ein Deep-Learning-Werkzeug, um die dreidimensionale Struktur der Pseudomonas-DNA-Gyrase zu analysieren und wahrscheinliche „Hotspots“ zu identifizieren — Cluster von Aminosäuren auf der Oberfläche, die für Bindungen besonders wichtig sind. Diese Regionen bilden die aktive Tasche des Enzyms, in der die normale DNA-Verarbeitung stattfindet und an der ein potenzieller Inhibitor idealerweise ansetzen würde.
Eine molekulare Begrüßung simulieren
Um zu prüfen, ob Im9 diese Hotspots erfassen kann, wandten sich die Forschenden molekularen Docking-Programmen zu, die Proteine virtuell wie 3D-Puzzle zusammenfügen. Zuerst bereinigten und vervollständigten sie die verfügbaren Strukturen von Im9 und der DNA-Gyrase, reparierten eine fehlende Schleife im Enzym und führten kurze Simulationen durch, um gespannte Bereiche zu entspannen. Anschließend nutzten sie zwei komplementäre Docking-Tools, ClusPro und LightDock, um viele Kandidatenkomplexe zu erzeugen. Aus diesen wählten sie die vielversprechendsten Anordnungen aus und unterzogen sie langen molekulardynamischen Simulationen von mehreren hundert Nanosekunden. Diese zeitaufgelösten „Filme“ erlaubten es, zu beobachten, wie sich die beiden Proteine anpassten, bogen und in stabilere Formen einpendelten, wenn sie miteinander verbunden waren.
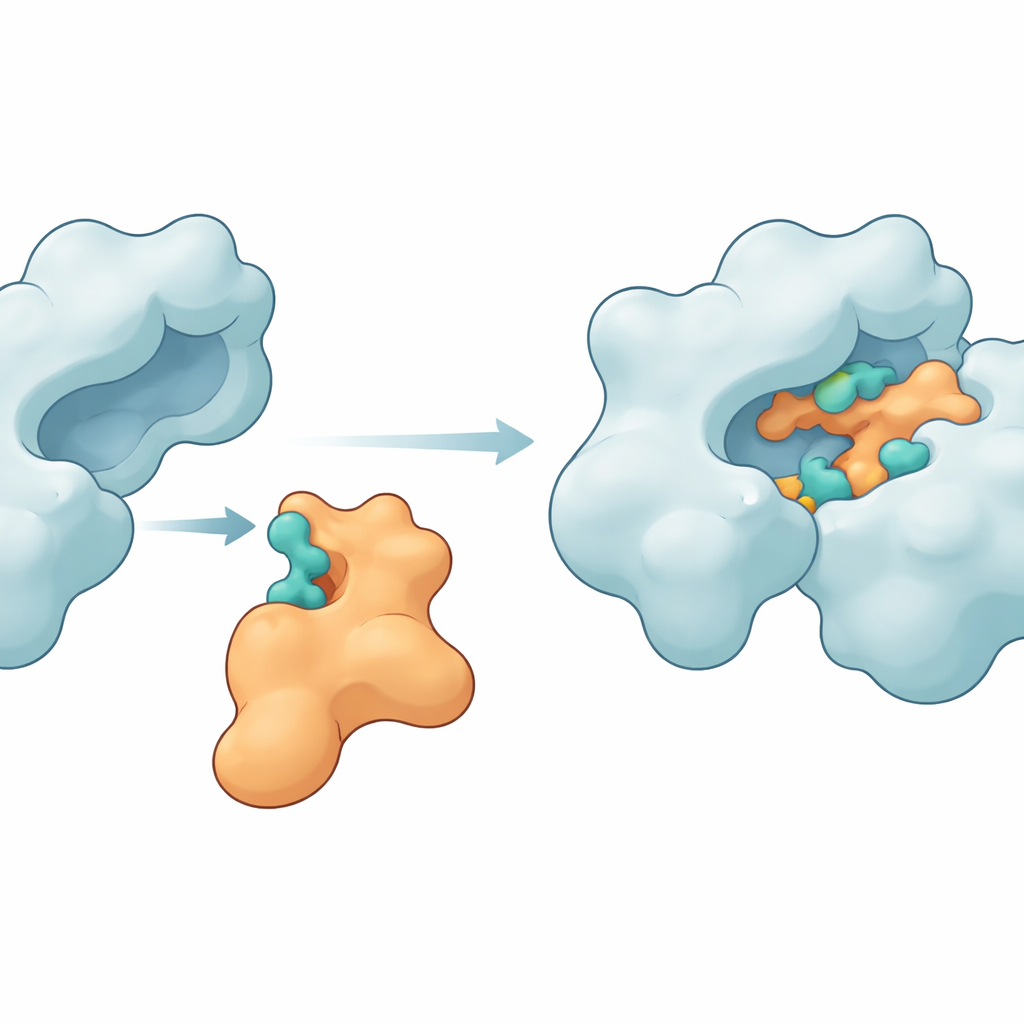
Wichtige Kontaktstellen, die die Proteine zusammenhalten
Die Simulationen zeigten, dass Im9 tatsächlich einen engen und beständigen Komplex mit der DNA-Gyrase bilden kann. Mehrere Aminosäuren am Enzym — wie MET27, ASP47, LYS105, LEU198, ASN199, ARG191 und GLU194 — bildeten wiederholt Wasserstoffbrücken und andere anziehende Wechselwirkungen mit entsprechenden Stellen auf Im9. In einem führenden Modell hielten die beiden Partner während eines Großteils der Simulation zwischen sechs und zehn Wasserstoffbrücken aufrecht, ein Hinweis auf eine starke und gut organisierte Schnittstelle. Weitere strukturelle Kenngrößen, einschließlich der Kompaktheit der Proteine und der Ausmaß ihrer Formschwankungen, zeigten, dass das Enzym intakt blieb, während das Immunitätsprotein gerade genug flexibel war, um sich an die Oberfläche der Gyrase anzupassen. Energieabschätzungen mithilfe der MM-GBSA-Methode stützten zusätzlich die Idee, dass diese Kontakte eine günstige, wenn auch moderate Bindungsfreie-Energie erzeugen, dominiert von elektrostatischen und van-der-Waals-Beiträgen.
Von Computermodellen zu künftigen Antimikrobiotika
Insgesamt deuten die Ergebnisse darauf hin, dass das Colicin E9-Immunitätsprotein stabil an die aktive Region der Pseudomonas-DNA-Gyrase binden kann und einen langlebigen Komplex bildet, der prinzipiell die normale Funktion des Enzyms bei der DNA-Verarbeitung blockieren könnte. Obwohl diese Befunde vollständig auf Computermodellen beruhen und noch experimentell überprüft werden müssen, liefern sie einen detaillierten Bauplan, wo und wie ein proteinbasierter Inhibitor ansetzen könnte. Für Nichtfachleute lautet die Kernbotschaft: Die Waffen und Schutzmechanismen der Naturbakterien können neue Strategien gegen schwer behandelbare Infektionen inspirieren. Durch das Verständnis dieses mikroskopischen „Handschlags“ auf atomarer Ebene rücken Wissenschaftler dem Ziel näher, neuartige Antimikrobiotika zu entwerfen, die entscheidende bakterielle Enzyme ausschalten, ohne menschliche Zellen zu schädigen.
Zitation: Alfaraj, R., Alkathiri, F. & Chikhale, R. Investigating Escherichia coli Colicin E9 immunity protein interactions with DNA gyrase of Pseudomonas aeruginosa: advanced computational approach for developing novel antimicrobial strategies. Sci Rep 16, 10786 (2026). https://doi.org/10.1038/s41598-026-44427-2
Schlüsselwörter: Antibiotikaresistenz, DNA-Gyrase, Protein–Protein-Wechselwirkungen, computationale Wirkstoffentwicklung, Pseudomonas aeruginosa