Clear Sky Science · de
Ab-initio-Bestimmung der Phasenstabilitäten dynamisch ungeordneter Feststoffe: rotatorische C2-Unordnung in Li2C2
Warum dieses wechselnde Feststoff wichtig ist
Viele moderne Technologien bauen auf Feststoffen auf, die ihre innere Struktur still verändern können, wenn sie erwärmt oder zusammengedrückt werden. Diese Änderungen, Phasenübergänge genannt, sind zentral für Konzepte wie Festkörperkühlung und sicherere Batterien. Die vorliegende Studie untersucht eine einfache Verbindung, Lithiumcarbid (Li2C2), die sich mit steigender Temperatur von einer ordentlich geordneten Form zu einer dynamisch unruhigeren, ungeordneten Form wandelt. Durch die Beobachtung dieser Transformation atomar in Computersimulationen zeigen die Autoren, wie die innere „Unruhe“ winziger molekularer Einheiten das Gleichgewicht zwischen zwei Kristallstrukturen verschieben kann.
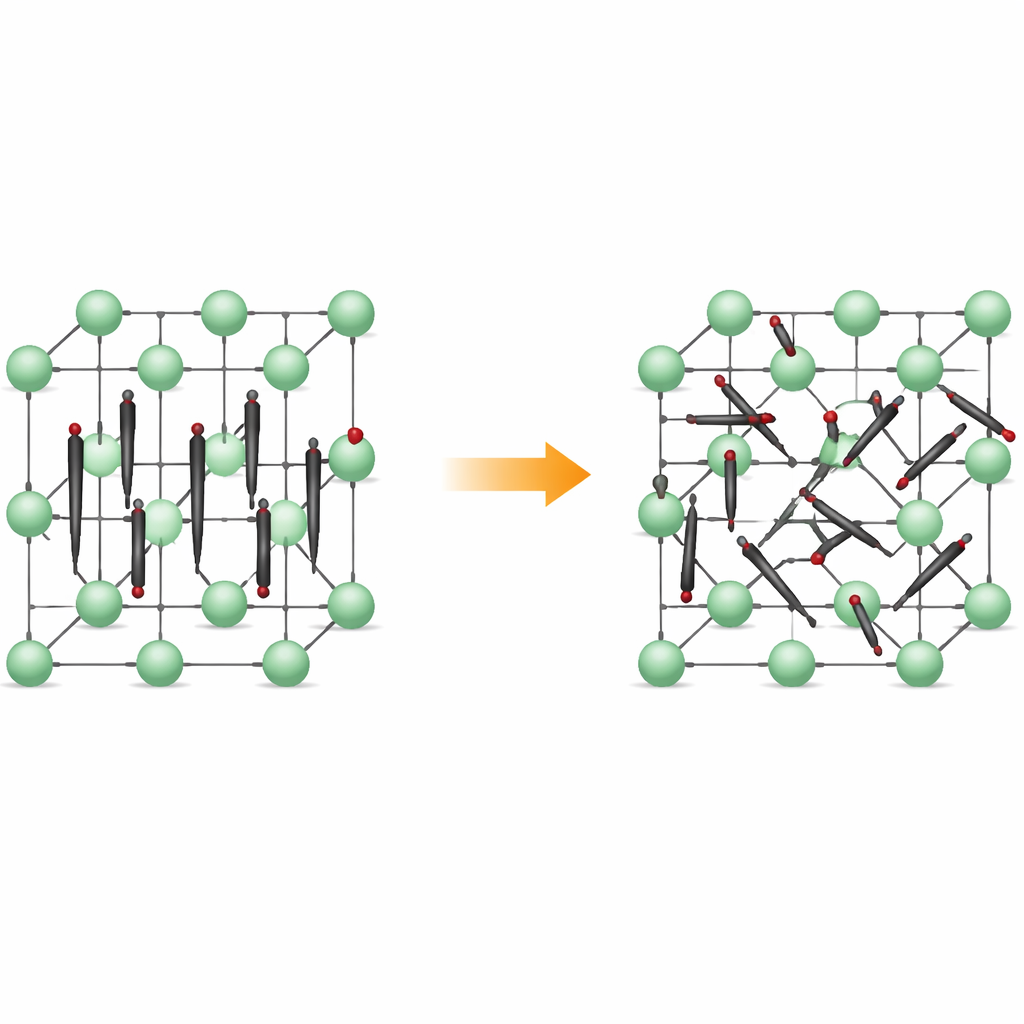
Von ordentlichen Reihen zu rastloser Bewegung
Bei niedrigen Temperaturen bildet Li2C2 ein orthorhombisches Kristallgitter: Die Kohlenstoffatome paaren sich zu kleinen C2-Dimeren, die alle in nahezu dieselbe Richtung zeigen, ähnlich ausgerichteter Streichhölzer. Lithium-Ionen sitzen dazwischen und bilden ein regelmäßiges dreidimensionales Gerüst. Beim Erhitzen wandelt sich das Material in eine kubische Form, in der die Zentren der Dimer auf einem Gitter geordnet bleiben, die Dimere selbst jedoch keine feste Orientierung mehr beibehalten. Stattdessen rotieren sie zwischen mehreren bevorzugten Orientierungen und verweilen in flachen Energiegruben, die bestimmten Ausrichtungen entsprechen. Das Material bleibt fest, doch seine innere Struktur wird dynamisch ungeordnet.
Die Veränderung entlang eines glatten Pfads verfolgen
Um zu verstehen, welche Phase bei einer bestimmten Temperatur stabiler ist, muss man ihre freien Energien vergleichen, die Energie und Entropie (ein Maß für Unordnung) kombinieren. Standardmethoden, die auf kleinen Schwingungen um feste Positionen beruhen, stoßen an ihre Grenzen, wenn Atome stark umherwandern oder rotieren. Die Autoren verwenden hier eine Technik namens Spannungs–Dehnungs-thermodynamische Integration, basierend auf ab-initio-Molekulardynamik. Sie konstruieren einen glatten Deformationspfad, der die Simulationszelle kontinuierlich von der tieftemperaturigen orthorhombischen Struktur in die hochtemperaturige kubische Form überführt. Entlang dieses Pfads führen sie lange Simulationen bei festen Temperaturen durch und messen, wie die innere Spannung auf die aufgezwungene Dehnung reagiert. Die Integration dieser Spannungsantwort liefert die freie Energie-Differenz zwischen den beiden Phasen.
Entropie durch atomare Bewegung sehen
Die Rechnungen zeigen, dass bei etwa 600 K die tieftemperaturige orthorhombische Phase noch leicht bevorzugt ist, während bei 650 K die kubische Phase um einige tausendstel Elektronenvolt pro Formeleinheit gewinnt. Interpoliert man zwischen diesen Ergebnissen, ergibt sich eine Übergangstemperatur von etwa 611 K. Das liegt unter experimentellen Schätzungen, ist aber angesichts der geringen Freienergieunterschiede noch in hinreichender Übereinstimmung. Die innere Energie der kubischen Phase ist tatsächlich höher; was sie stabilisiert, ist ein großer Entropiegewinn, der direkt auf die rotatorische Unordnung der C2-Dimere zurückgeführt wird. Durch die Analyse, wie die Orientierung jedes Dimers ihre Anfangsrichtung im Laufe der Zeit verliert, zeigen die Autoren, dass sich die Dimere auf Sub-Pikosekunden-Zeitskalen neu orientieren und damit die Grenze zwischen den üblichen Kategorien „vibrationsbedingte“ und „konfigurationsbedingte“ Entropie verwischen.
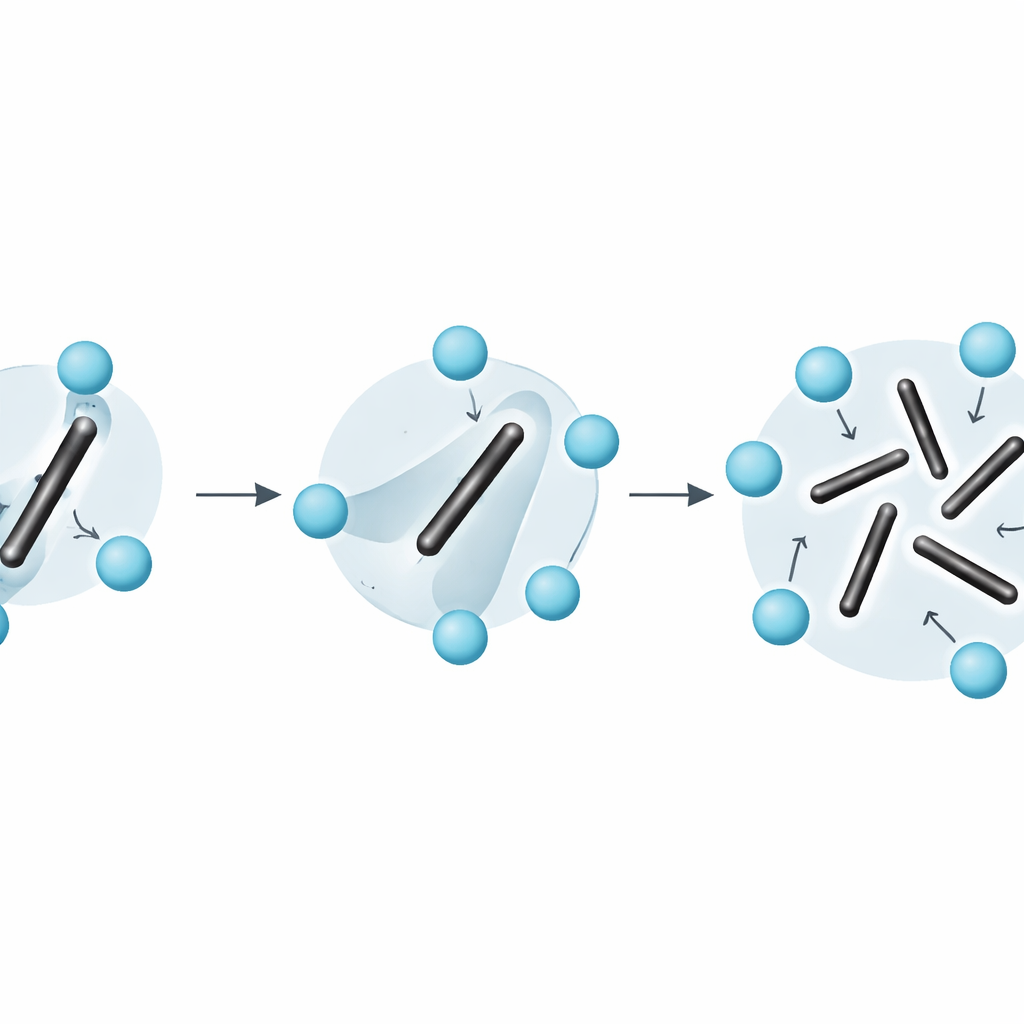
Jenseits einfacher Vorstellungen von Festkörperunordnung
Die Arbeit macht außerdem deutlich, dass gängige Vereinfachungen — etwa die Behandlung der Entropie als einfache Summe aus Schwingungen um feste Konfigurationen plus separate Zählung statischer Orientierungen — für Materialien wie Li2C2 versagen. Weil die Dimerrotationen schnell sind und stark mit gewöhnlichen Schwingungen gekoppelt sind, lässt sich das System nicht sauber in getrennte „schwingende“ und „umordnende“ Teile zerlegen. Die Spannungs–Dehnungs-Integrationsmethode umgeht dieses Problem: Sie extrahiert die vollständige freie Energie direkt aus der mikroskopischen Dynamik, ohne dass man raten muss, wie die Entropie aufzuteilen ist.
Was uns die Studie lehrt
Alltäglich ausgedrückt zeigt die Studie, wie ein Feststoff starr bleiben kann, während sich seine inneren Bausteine zunehmend frei drehen und wenden, und wie diese innere Freiheit eine stärker ungeordnete Struktur thermodynamisch bevorzugt machen kann. Für Li2C2 wird die hochtemperaturige kubische Phase nicht dadurch stabilisiert, dass sie energetisch günstiger wäre, sondern weil sie viel mehr Möglichkeiten bietet, wie sich die C2-Dimere orientieren und bewegen können. Indem gezeigt wird, dass die Spannungs–Dehnungs-thermodynamische Integration dieses feine Gleichgewicht zwischen Ordnung, Energie und Entropie erfassen kann, eröffnet die Arbeit einen Weg zur Vorhersage ähnlicher Übergänge in anderen dynamisch ungeordneten Feststoffen, die zukünftige Kühlgeräte, Batterien und intelligente Materialien untermauern könnten.
Zitation: Klarbring, J., Filippov, S., Häussermann, U. et al. Ab initio determination of phase stabilities of dynamically disordered solids: rotational C2 disorder in Li2C2. Sci Rep 16, 8965 (2026). https://doi.org/10.1038/s41598-026-43795-z
Schlüsselwörter: Festkörperphasenübergang, dynamische Unordnung, Molekulardynamik, Lithiumcarbide, thermodynamische Integration