Clear Sky Science · de
Hybrid-optimiertes Deep-Learning-Modell zur Diagnose von Brustkrebs anhand genetischer Daten
Warum das für Patientinnen und Familien wichtig ist
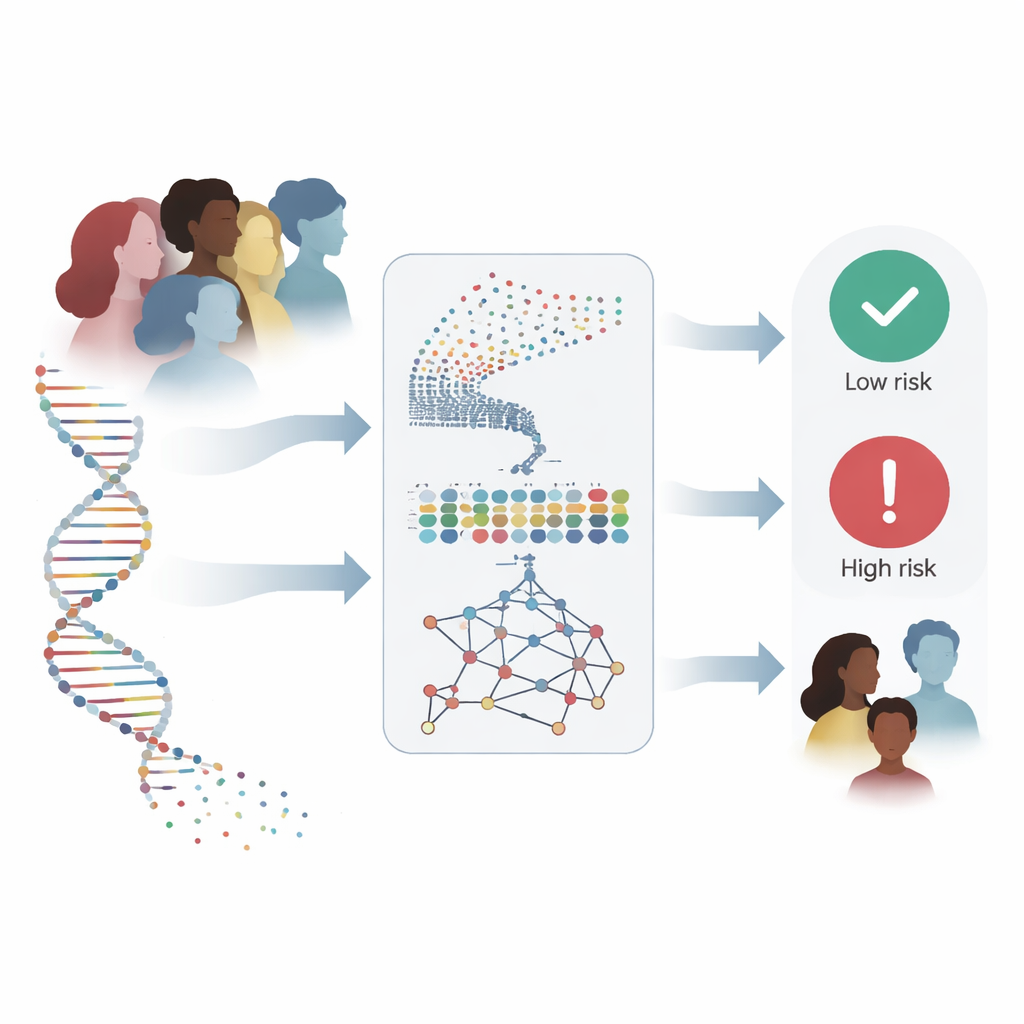
Brustkrebs ist heute weltweit die am häufigsten diagnostizierte Krebserkrankung bei Frauen, und eine frühe Erkennung kann über Leben und Tod entscheiden. Ärztinnen und Ärzte haben zunehmend Zugang zu genetischen Informationen, doch aus Zehntausenden Genmessungen klare Antworten zu gewinnen, ist außerordentlich schwierig. Dieser Artikel beschreibt ein neues Computermodell, das diese komplexen genetischen Muster liest, um Brustkrebs zu erkennen und Prognosen mit auffallender Genauigkeit zu stellen — potenziell ein leistungsfähiger Assistent für frühere und verlässlichere klinische Entscheidungen.
Von Genen zu Warnzeichen
Jeder Brusttumor trägt einen molekularen Fingerabdruck, der in der Aktivität tausender Gene kodiert ist. Die Autorinnen und Autoren wollten ein System entwickeln, das diesen Fingerabdruck direkt liest, anstatt sich nur auf Bilder oder eine Handvoll bekannter Gene wie BRCA1 und BRCA2 zu stützen. Sie arbeiteten mit zwei der größten öffentlichen Ressourcen der Krebsgenomik: dem TCGA-Brustkrebs-Kohorten-Datensatz, der Genaktivitäten für 17.814 Gene in 590 Proben enthält, und der METABRIC-Studie, die genomische und klinische Informationen von mehr als 1.400 Patientinnen umfasst. Ihr Ziel war ehrgeizig: eine Methode zu entwerfen, die mit dieser Datenflut umgehen, die aussagekräftigsten Signale finden und zuverlässig in völlig unabhängigen Patientengruppen funktionieren kann.
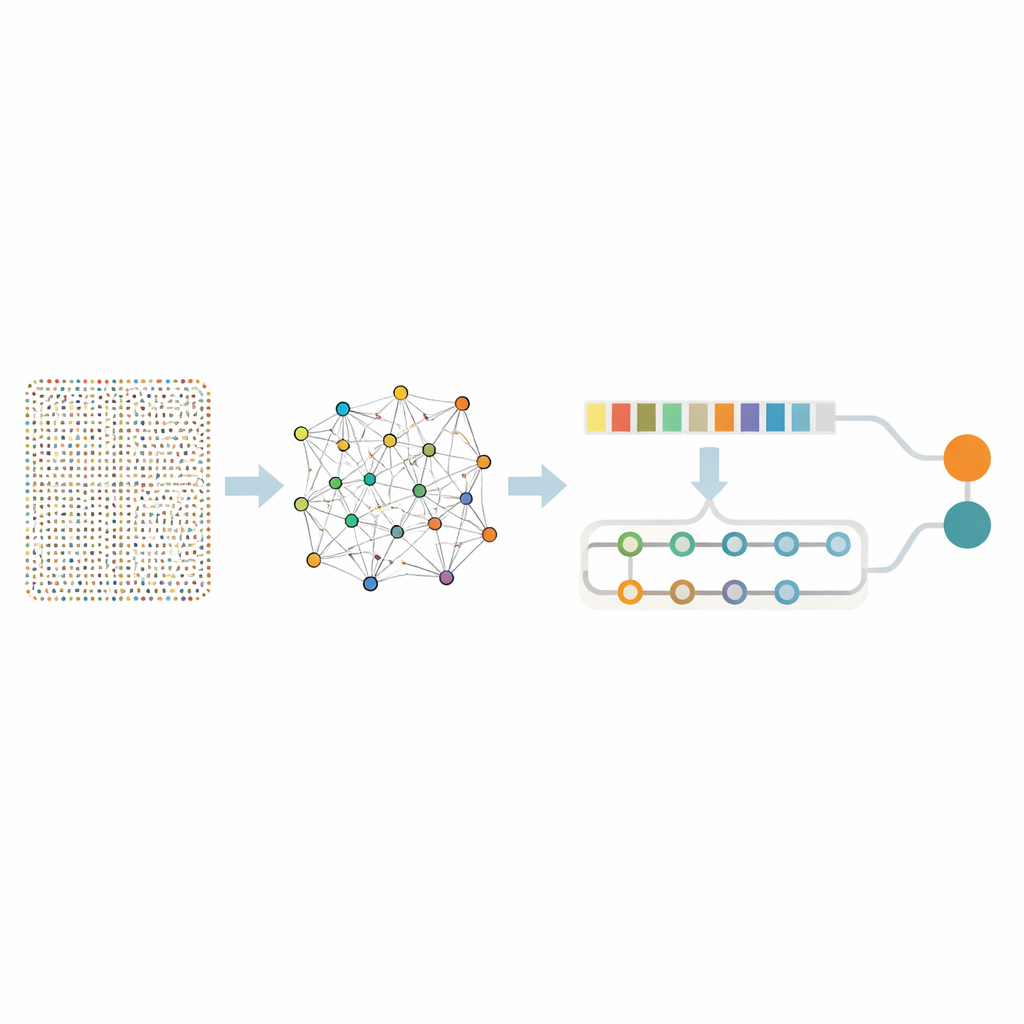
Tausende Gene auf eine nützliche Menge reduzieren
Fast achtzehntausend Gene gleichzeitig zu betrachten, ist selbst für fortgeschrittene Algorithmen überwältigend und birgt das Risiko, bedeutungslose Rauschsignale zu erfassen. Die Forschenden verwendeten daher ein zweistufiges "Sieb", um eine kleinere Menge wirklich informativer Gene zu isolieren. Zunächst wandten sie eine Technik namens Random Forest an, die praktisch viele Entscheidungsbäume befragt, welche Gene am wichtigsten sind, um krankes Gewebe von gesundem zu unterscheiden. Dieser Schritt reduzierte die Liste auf 436 vielversprechende Gene. Anschließend untersuchten sie, wie diese Gene gemeinsam agieren, mittels Association-Rule-Mining — einer Methode, die Gruppen von Genen erkennt, die in Tumoren tendenziell gleichzeitig aktiv sind. Diese zusätzliche Analyseebene identifizierte Genpaare und Netzwerke, die mit zentralen Krebsprozessen wie schneller Zellteilung, DNA-Schadensreparatur und Veränderungen im umgebenden Gewebe verbunden sind. Nach dieser Verfeinerung blieben 332 Gene übrig — biologisch reich an Bedeutung, aber weitaus handhabbarer für tiefere Analysen.
Ein zweiteiliges neuronales Netzwerk, das Muster und Kontext lernt
Mit diesem fokussierten Gen-Set baute das Team ein hybrides Deep-Learning-Modell, das zwei Arten neuronaler Netze kombiniert. Ein Teil, bekannt als Faltungsnetzwerk (Convolutional Network), scannt entlang der Genliste, um lokale Muster aufzunehmen — Cluster von Genen, die dazu neigen, gemeinsam anzusteigen oder abzufallen. Der zweite Teil, ein bidirektionales Speicher-Netzwerk, betrachtet dieselben Informationen unter Wahrung langfristiger Beziehungen und erfasst, wie entfernte Gene einander über das gesamte Profil hinweg beeinflussen. Vor dem Training balancierten die Autorinnen und Autoren die Daten so, dass Krebs- und Nicht-Krebs-Proben fair vertreten waren, und fügten kleine Mengen künstlichen Rauschens hinzu, um dem Modell beizubringen, sich nicht von zufälligen Schwankungen täuschen zu lassen.
Wie gut das System in Tests unter realen Bedingungen abschneidet
Beim Training und Testen auf TCGA-Daten konnte das hybride Netzwerk Tumor- von Normalproben mit etwa 97 % Genauigkeit korrekt unterscheiden und zeigte eine nahezu perfekte Trennschärfe zwischen beiden Gruppen. Wichtig ist, dass es einfachere Deep-Learning-Setups und gängige Machine-Learning-Methoden wie logistische Regression und Support-Vektor-Maschinen übertraf, selbst wenn diese Konkurrenzmethoden dieselben sorgfältig ausgewählten Gene erhielten. Die stärkere Prüfung bestand jedoch darin, ob das Modell auf einem völlig anderen Datensatz standhält. Auf METABRIC angewendet, das in anderen Krankenhäusern mit unterschiedlichen labortechnischen Methoden erhoben wurde, behielt das System eine hohe Leistung bei: In seinem besten Lauf erreichte es 99,3 % Genauigkeit und identifizierte korrekt jede Patientin, die später an Brustkrebs starb — eine entscheidende Eigenschaft, wenn das Werkzeug dazu dienen soll, Hochrisikofälle zu kennzeichnen.
Was das für die künftige Versorgung bedeuten könnte
Für Nichtfachleute lautet die Quintessenz, dass diese Studie einen intelligenten Filter und Leser für genetische Daten liefert, der Brustkrebs und damit verbundene Risiken mit bemerkenswerter Konsistenz in großen Patientengruppen erkennen kann. Durch die Kombination einer durchdachten Gen-Auswahlstrategie mit einem zweiteiligen neuronalen Netzwerk zeigen die Autorinnen und Autoren, dass Computer klinisch bedeutsame Signale aus enormen genetischen Datensätzen extrahieren können — nicht nur in einer Studie, sondern über unabhängige Kohorten hinweg. Zwar sind weitere Arbeiten nötig, um den Ansatz in vielfältigen Populationen zu prüfen und seine Entscheidungen detailliert zu erklären, doch weist die Methode in Richtung einer Zukunft, in der eine einfache Blut- oder Gewebeprobe in solche Modelle eingespeist werden könnte, um Ärztinnen und Ärzten zu helfen, Tumoren früher zu erkennen und Behandlungen präziser zuzuschneiden.
Zitation: Hesham, F., Abbassy, M.M. & Abdalla, M. Hybrid tuned deep learning model for breast cancer diagnosis using genetic data. Sci Rep 16, 9664 (2026). https://doi.org/10.1038/s41598-026-41643-8
Schlüsselwörter: Brustkrebs-Genomik, Deep-Learning-Diagnose, Genexpressions-Biomarker, früherkennung von Krebs, klinische Entscheidungsunterstützung