Clear Sky Science · de
Entdeckung von Hydroxytriazol als potenzieller Glyoxalase‑I‑Inhibitor mithilfe rechnergestützter Wirkstoffdesign‑Methoden
Warum das Ausschalten eines winzigen Zellreinigers Krebs bekämpfen könnte
Krebszellen wachsen oft so schnell, dass sie in ihren eigenen Abfallstoffen ersticken. Ein Überlebenskniff ist eine eingebaute Aufräumtruppe, die schädliche Nebenprodukte des Zuckerabbaus entgiftet. Diese Studie untersucht, wie sich ein Schlüsselmitglied dieser Mannschaft, das Enzym Glyoxalase‑I, ausschalten lässt, indem Computer genutzt werden, um Zehntausende von Molekülen zu durchsieben, und Experimente die besten Kandidaten testen. Ziel ist es, neue Wirkstoff‑„Startpunkte“ zu finden, die eines Tages Ärzten helfen könnten, Krebszellen von innen gezielt zu vergiften.
Ein verborgenes Abfallsystem in unseren Zellen
Jede Zelle baut ständig Zucker ab, um Energie zu gewinnen, und dabei entsteht ein reaktives Abfallprodukt namens Methylglyoxal. In normalen Mengen wandelt unser Körper Methylglyoxal über das Glyoxalase‑System — einen Zweischrittweg, der auf dem Hilfsmolekül Glutathion beruht — in unschädliche Milchsäure um. Glyoxalase‑I ist der erste und wichtigste Schritt dieser Kette. Krebszellen, die Zucker im Eiltempo verbrennen, sind stark auf Glyoxalase‑I angewiesen, um zu verhindern, dass Methylglyoxal toxische Level erreicht. Wird dieses Enzym blockiert, baut sich Methylglyoxal auf und kann geschädigte Zellen in den programmierten Zelltod treiben. Das macht Glyoxalase‑I zu einem attraktiven Ziel für krebsbekämpfende Wirkstoffe, die eine grundlegende Schwäche des Tumorstoffwechsels ausnutzen.
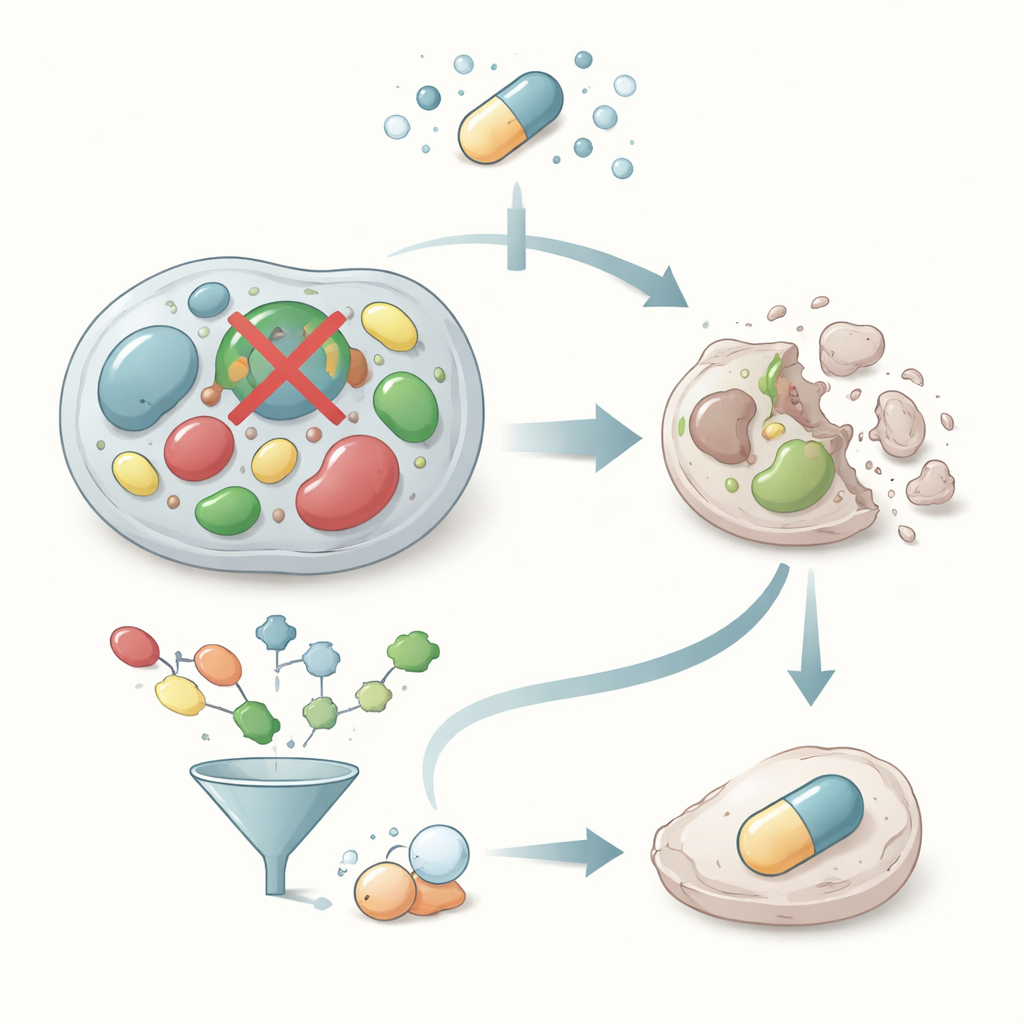
Chemischen Raum mit Silizium und Statistik durchkämmen
Anstatt im Labor zufällige Substanzen zu testen, nutzten die Forscher rechnergestütztes Wirkstoffdesign, um eine große kommerzielle Sammlung von mehr als 50.000 kleinen Molekülen zu durchsuchen. Spezialisierte Software bereinigte und standardisierte zunächst jedes Molekül und sagte dann seine 3D‑Gestalt und sein Verhalten bei körperähnlichem pH‑Wert voraus. Ein schnelles virtuelles Screening bewertete, wie gut jeder Kandidat in die aktive Stelle von Glyoxalase‑I passen könnte. Das Team wendete dann einfache Regeln zu Größe, Löslichkeit und anderen wirkstoffähnlichen Eigenschaften an, um Moleküle auszuschließen, die im Körper wahrscheinlich nicht funktionieren würden. Ein detaillierteres Docking‑Programm untersuchte, wie sich die vielversprechendsten Moleküle innerhalb des Enzyms ausrichten könnten — insbesondere, wie sie das Zink‑Metallatom erreichen und fassen könnten, das im Zentrum der Glyoxalase‑I‑Chemie sitzt.
Ein neuer Weg, den metallenen Kern des Enzyms zu greifen
Frühere Versuche, Glyoxalase‑I zu blockieren, konzentrierten sich auf bekannte chemische Gruppen wie Carbonsäuren und Hydroxamsäuren, die gut Metall binden, aber oft unter schlechter Stabilität oder unerwünschten Nebenwirkungen leiden. Die vorliegende Studie identifizierte stattdessen eine andere Art von „Metallgreifer“: einen Hydroxytriazolring. Unter sechzehn hochrangigen Molekülen, die zum Kauf und zur Labortestung ausgewählt wurden, stach eines mit diesem Ring — codiert SPB07393SC — deutlich hervor. Im virtuellen Docking reichte seine Hydroxytriazol‑Gruppe bis zum Zinkatom, während seine zwei aromatischen Ringe in nahegelegene lipophile Taschen des Enzyms schlüpften. Computersimulationen des Komplexes über einige zehn Nanosekunden deuteten darauf hin, dass das Molekül fest gebunden blieb, mit stabilen Abständen, kompakter Proteinstruktur und einem beständigen Netzwerk von Wasserstoffbrücken.
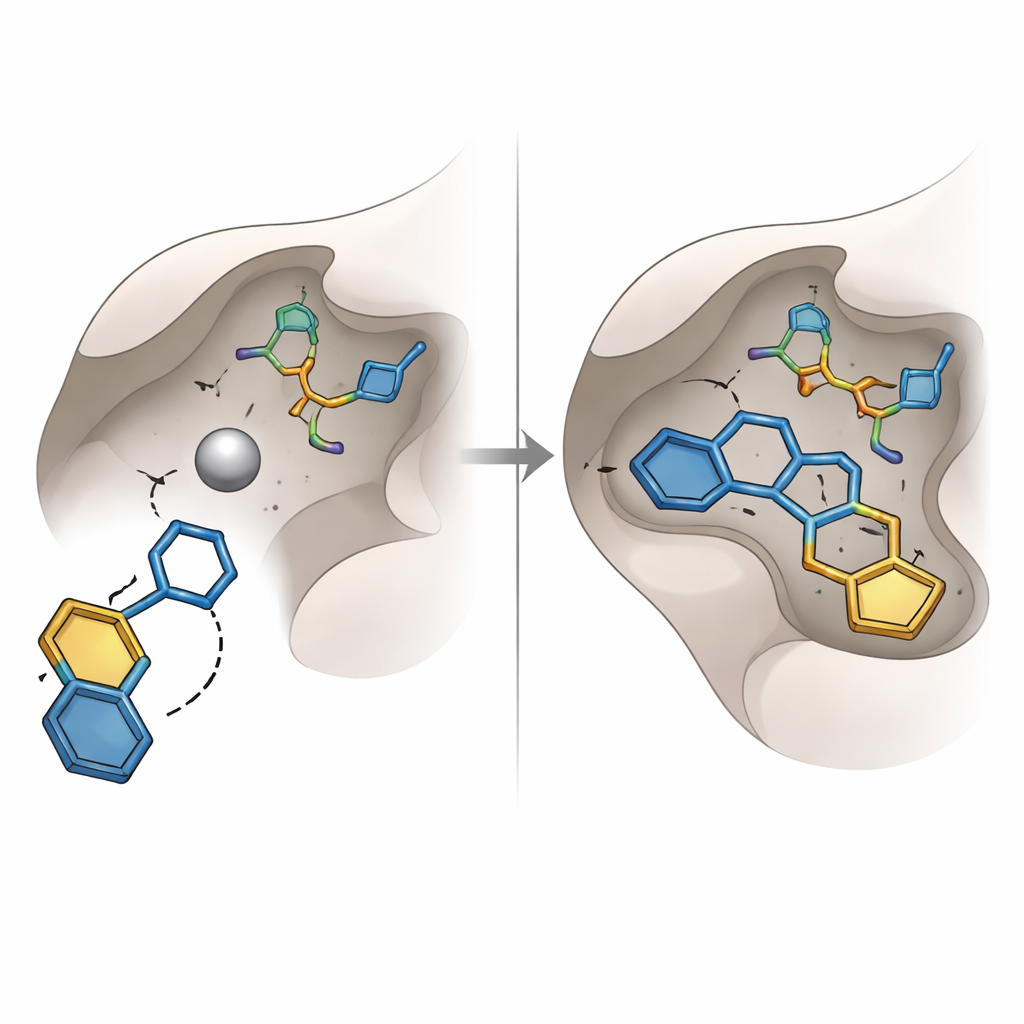
Die Vorhersagen im Test
Um zu prüfen, ob die Computermodelle reale Effekte vorhersagten, maßen die Forscher, wie gut die ausgewählten Moleküle die Aktivität von gereinigtem menschlichem Glyoxalase‑I in einem Plattenassay verlangsamten. Fünfzehn der sechzehn Kandidaten zeigten unter den getesteten Bedingungen nur schwache oder vernachlässigbare Hemmung, was die Fallstricke des alleinigen Verlassens auf statische Docking‑Werte unterstreicht. Im Gegensatz dazu hemmte SPB07393SC das Enzym stark, mit einer mittleren Mikromolar‑Potenz, die es eher zu einem soliden frühen „Hit“ als zu einem fertigen Wirkstoff macht. Zusätzliche Software‑Tools sagten voraus, dass dieses Molekül eine akzeptable Löslichkeit, gute Resorption, die Möglichkeit, bei Bedarf das Gehirn zu erreichen, und ein geringes Risiko für bestimmte genetische oder leberbezogene Toxizitäten haben sollte — obwohl diese Sicherheitsvorhersagen noch experimentell bestätigt werden müssen.
Was das für zukünftige Krebsmedikamente bedeutet
Die Arbeit führt Hydroxytriazol als neue Möglichkeit ein, Wirkstoffkandidaten am Zinkatom im Kern von Glyoxalase‑I zu verankern, und erweitert damit die chemischen Werkzeuge, die Wirkstoffdesigner zur Verfügung stehen. Während SPB07393SC selbst nur ein Ausgangspunkt ist, kennzeichnen seine Kombination aus Enzymblockade, vorhergesagtem wirkstoffähnlichem Verhalten und stabiler Bindung in bewegungsbasierten Simulationen es als vielversprechendes Gerüst für weitere Optimierungen. Allgemeiner zeigt die Studie sowohl die Stärken als auch die Grenzen rechnergestützter Suche: Sie kann große chemische Bibliotheken schnell auf einige realistische Anwärter eingrenzen, doch sorgfältige Laborversuche bleiben unerlässlich, um zu erkennen, welche Moleküle das Enzym tatsächlich deaktivieren, auf das Krebszellen zur Entsorgung ihrer toxischen Abfälle angewiesen sind.
Zitation: Al-Qazzan, M., Al-Balas, Q., Alnajjar, B. et al. Discovery of hydroxytriazole as a potential glyoxalase-I inhibitor utilizing computer-aided drug design techniques. Sci Rep 16, 9945 (2026). https://doi.org/10.1038/s41598-026-40497-4
Schlüsselwörter: glyoxalase I, Krebsstoffwechsel, rechnergestütztes Wirkstoffdesign, Zink‑bindende Inhibitoren, molekulares Docking