Clear Sky Science · de
DPAS: Disease-Associated Peptide Anomaly Score zur Identifizierung pathogener Peptide mittels One-Class-Learning
Warum winzige Proteinabschnitte für unsere Gesundheit wichtig sind
Peptide — kurze Abschnitte von Proteinen — haben sich zu wichtigen Akteuren in der modernen Medizin entwickelt. Sie können als präzise Botenstoffe im Körper fungieren und werden zunehmend als Wirkstoffe und Krankheitsmarker genutzt. Doch herauszufinden, welche Peptide tatsächlich mit Krankheiten verbunden sind, setzt meist eindeutige Beispiele sowohl für „krankheitsassoziierte“ als auch für „nicht‑krankheitsassoziierte“ Peptide voraus — etwas, das die Biologie selten liefert. Diese Studie stellt einen neuen Ansatz vor, um potenziell schädliche Peptide allein anhand der bereits bekannten krankheitsrelevanten Beispiele zu erkennen und so einen schnelleren und weniger verzerrten Weg zur Entdeckung künftiger Diagnostika und Therapien zu eröffnen.
Die Herausforderung bei der Bestimmung der „Nicht‑Krankheits“-Gruppe
Traditionelle Computermodelle lernen durch den Vergleich zweier Seiten: positive Beispiele, die als krankheitsrelevant bekannt sind, und negative Beispiele, die als harmlos gelten. In der Peptidforschung ist diese zweite Gruppe problematisch. Viele Peptide wurden schlichtweg nicht getestet, sodass ihre Kennzeichnung als „nicht‑krankheitsrelevant“ irreführend sein und Verzerrungen einführen kann. Frühere Studien zu anti‑krebserregenden oder anti‑entzündlichen Peptiden erzielten zwar beeindruckende Genauigkeiten, beruhten jedoch häufig auf manuell erstellten oder vermuteten negativen Datensätzen. Folglich können deren Modelle bei seltenen Signalen oder neuen Arten von krankheitsrelevanten Peptiden, die sich vom Trainingsdatensatz unterscheiden, Schwierigkeiten haben.
Aus dem lernen, was wir wissen, statt aus dem, was wir vermuten
Die Autoren wählen einen anderen Weg: Anstatt ein zweiseitiges Problem zu erzwingen, betrachten sie krankheitsassoziierte Peptide als eine kohärente Gruppe und fragen: „Wie sieht diese Gruppe im Detail aus?“ Sie sammeln über 760.000 mutierte menschliche Peptide aus einer spezialisierten, krebsbezogenen Datenbank und beschreiben jedes Peptid durch einen reichhaltigen Satz an Merkmalen. Dazu gehören Häufigkeiten einzelner Aminosäuren, die Anordnung von Aminosäurepaaren, grundlegende physikalisch‑chemische Eigenschaften wie Volumen und Hydrophilie sowie kurz wiederkehrende Sequenzmuster, sogenannte Motive. Eine Technik namens Hauptkomponentenanalyse (PCA) komprimiert dann diese hochdimensionale Beschreibung in eine handlichere Form, wobei die wichtigsten Variationsquellen erhalten bleiben.
Ungewöhnliche Peptide mit One‑Class‑Modellen erkennen
Mit diesem komprimierten Merkmalsraum trainiert das Team drei „One‑Class“-Modelle — Algorithmen, die die Gestalt einer einzelnen Gruppe erlernen und alles, was nicht passt, markieren. Sie testen One‑Class Support Vector Machines, Isolation Forests und eine Art neuronales Netzwerk namens Autoencoder. Der Autoencoder lernt, die Merkmale jedes Peptids auf eine schmale interne Darstellung zu verdichten und anschließend zu rekonstruieren; Peptide, die dem gelernten Krankheitsmuster entsprechen, werden genau wiederhergestellt, während ungewöhnliche Peptide einen höheren Rekonstruktionsfehler verursachen. Der Vergleich normalisierter Anomaliewerte über alle Methoden zeigt, dass der Autoencoder den engsten Cluster typischer Peptide und die klarste Trennung zwischen Inliern und Ausreißern liefert. Durch das Setzen einer Schwelle beim Rekonstruktionsfehler um das 95. Perzentil klassifiziert das Modell die Mehrzahl der Peptide als wahrscheinlich krankheitsassoziiert und markiert beständig einen kleinen Bruchteil als atypisch.
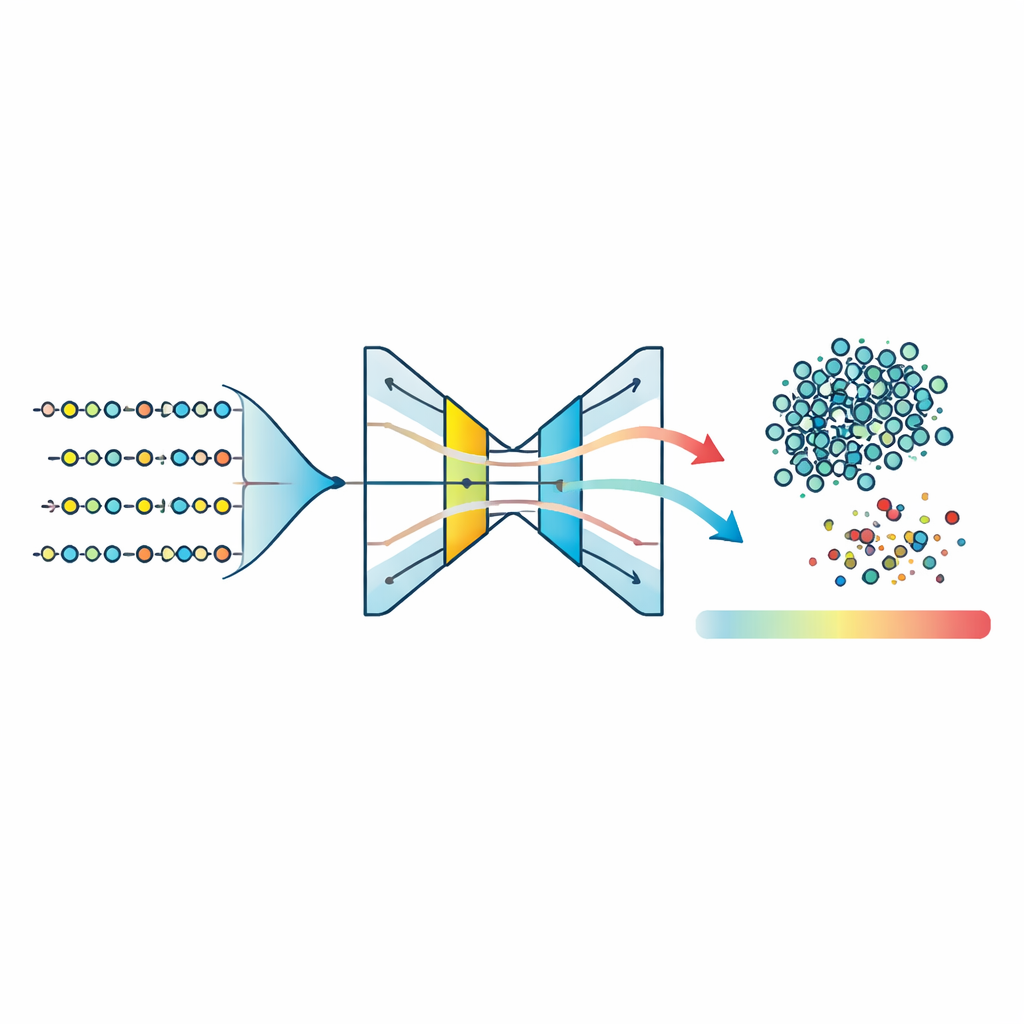
Komplexe Werte in eine einzige, aussagekräftige Zahl überführen
Um die Ergebnisse biologisch besser interpretierbar zu machen, führen die Autoren den Disease Peptide Anomaly Score (DPAS) ein. Dieser Score vereint zwei Komponenten: wie ungewöhnlich ein Peptid für den Autoencoder erscheint (sein normalisierter Rekonstruktionsfehler) und wie stark dessen Merkmale zu Vorhersagen beitragen, gemessen mit einer gängigen Erklärmethode namens SHAP. In der Praxis erweisen sich Motive und spezifische physikalisch‑chemische Eigenschaften als besonders aussagekräftig. DPAS kombiniert diese Signale so, dass Peptide, die sowohl strukturell auffällig sind als auch durch biologisch sinnvolle Merkmale gestützt werden, höhere Ränge erhalten. Die am höchsten bewerteten Peptide werden anschließend mit einem Motiv‑Suchwerkzeug untersucht, das sie mit bekannten funktionellen Signaturen wie Phosphorylierungsstellen, Metallbindungsregionen und anderen regulatorischen Mustern verknüpft, die häufig an Signalübertragung und Enzymsteuerung beteiligt sind.
Was das für zukünftige Diagnostik und Medikamente bedeutet
Vereinfacht gesagt bietet diese Arbeit einen intelligenteren Filter, um verdächtige Peptide zu finden, ohne vorzutäuschen, welche eindeutig harmlos sind. Indem nur aus bestätigten krankheitsrelevanten Beispielen gelernt und neue Kandidaten mit DPAS priorisiert werden, können Forschende eine kurze, biologisch plausible Liste von Peptiden für Labortests erstellen. Viele der höchstbewerteten Kandidaten enthalten bekannte funktionelle Motive, was die Annahme stützt, dass sie an Krankheitsprozessen beteiligt sein könnten. Obwohl die Methode weiterhin Annahmen voraussetzt und experimentell nachgewiesene „sichere“ Peptide für eine vollständige Validierung fehlen, liefert sie eine realistischere und transparentere Grundlage für die Entdeckung peptidbasierter Biomarker und könnte auf andere biologische Datentypen angepasst werden, bei denen verlässliche negative Beispiele knapp sind.
Zitation: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
Schlüsselwörter: krankheitsassoziierte Peptide, Anomalieerkennung, Autoencoder, Biomarker-Entdeckung, One-Class-Learning