Clear Sky Science · de
De-novo-Generierung und In-silico-Screening von antidiabetischen Peptidkandidaten mittels eines Deep-Learning–Attention-Frameworks mit Verschmelzung physikochemischer Merkmale
Warum intelligenteres Peptiddesign bei Diabetes wichtig ist
Diabetes betrifft weltweit Hunderte Millionen Menschen, und die aktuellen Medikamente wirken nicht bei allen Patienten gleich gut. Viele Behandlungen verlieren mit der Zeit an Wirksamkeit oder verursachen Nebenwirkungen. Eine vielversprechende neue Option sind kleine Proteine, sogenannte antidiabetische Peptide, die den Blutzucker mit hoher Präzision feinsteuern können. Die Herausforderung besteht darin, dass die Suche nach neuen Peptidwirkstoffen im Labor langsam und teuer ist. Diese Studie stellt eine computergestützte Pipeline vor, die große Mengen potenzieller antidiabetischer Peptide erzeugen und durchmustern kann, um Forschende zu den vielversprechendsten Kandidaten für die experimentelle Prüfung zu führen.
Von bekannten Diabetes-Peptiden zu sauberen Ausgangsdaten
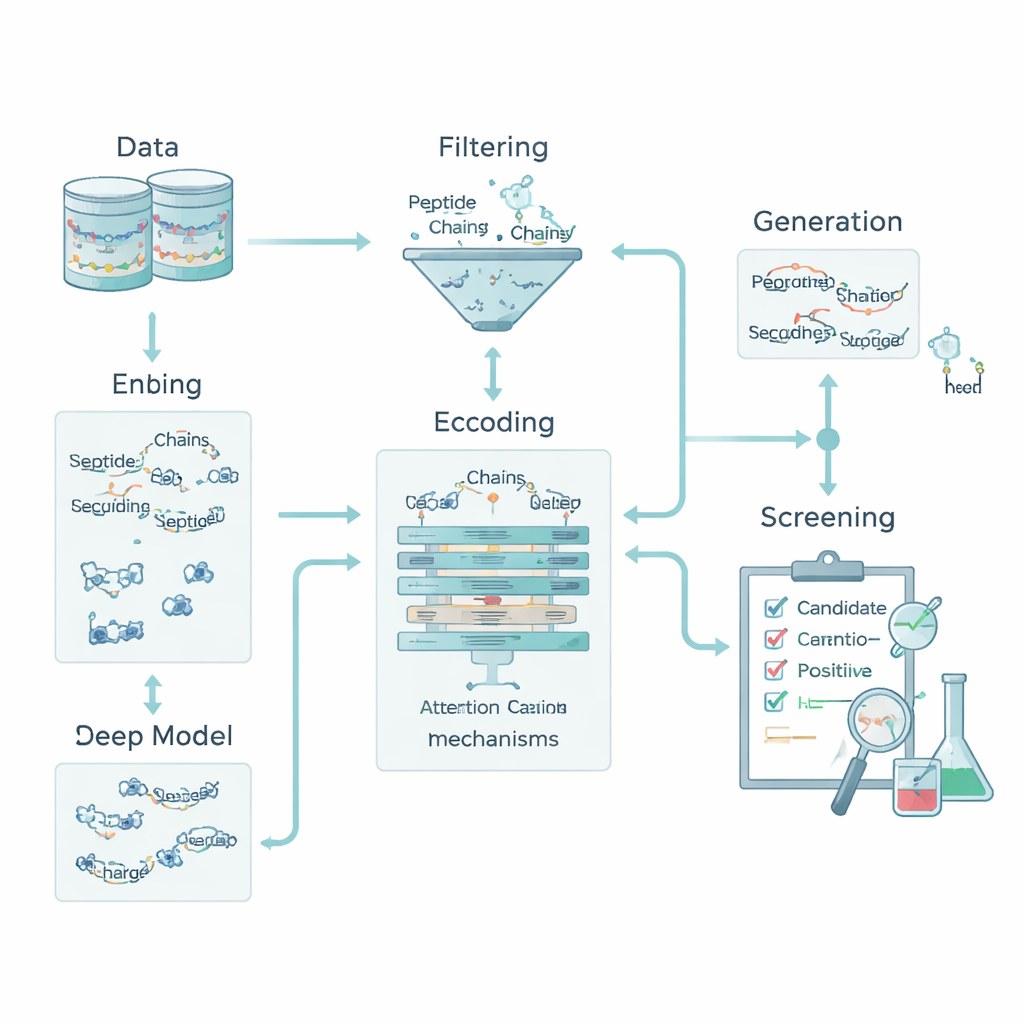
Die Forschenden begannen mit dem Zusammenstellen einer qualitativ hochwertigen Sammlung von Peptiden, die experimentell gezeigt haben, den Blutzucker zu beeinflussen, meist über Hormone wie GLP‑1 oder Enzyme wie DPP‑IV. Diese bildeten die „positiven“ Beispiele. Daraus bauten sie ein korrespondierendes „negatives“ Set aus Peptiden ohne gemeldete antidiabetische Aktivität, sorgfältig ausgewählt, sodass Länge, Zusammensetzung und grundlegende Chemie den positiven ähnlich waren. Um zu vermeiden, dass das Modell durch nahezu identische Sequenzen getäuscht wird, nutzten sie Sequenzähnlichkeits-Tools, um sicherzustellen, dass eng verwandte Peptide niemals gleichzeitig in Trainings- und Testgruppen vorkamen. Diese homologie-bewusste Aufteilung stellte sicher, dass das System anhand seiner Fähigkeit bewertet wird, wirklich neue Muster zu erkennen, statt alte auswendig zu lernen.
Chemie kodieren, damit Maschinen Peptide lesen können
Für einen Computer ist ein Peptid nur eine Buchstabenfolge, die Aminosäuren repräsentiert. Um diese Buchstaben mit biologischer Bedeutung zu verknüpfen, wandelte das Team jede Aminosäure in fünf grundlegende chemische Merkmale um: Hydrophobizität (Wasserabstoßung), elektrische Ladung, Neigung zur Wasserstoffbrückenbildung, Masse und das Vorhandensein eines aromatischen Rings. So wurde jedes Peptid zu einem kleinen „Bild“, das sowohl Reihenfolge als auch Chemie einfängt. Darüber hinaus fügten sie Peptid-weitere Deskriptoren hinzu, wie Gesamtladung, mittlere Hydrophobizität und den Boman‑Index, der mit der Neigung eines Peptids zur Bindung an andere Proteine zusammenhängt. Zusammen erlauben diese Merkmale dem Modell, sowohl lokale Muster – kurze Aminosäure‑Motife – als auch globale Eigenschaften zu betrachten, die das Verhalten eines Peptids im Körper beeinflussen.
Eine Deep-Learning‑Engine, die ihre Entscheidungen erklärt
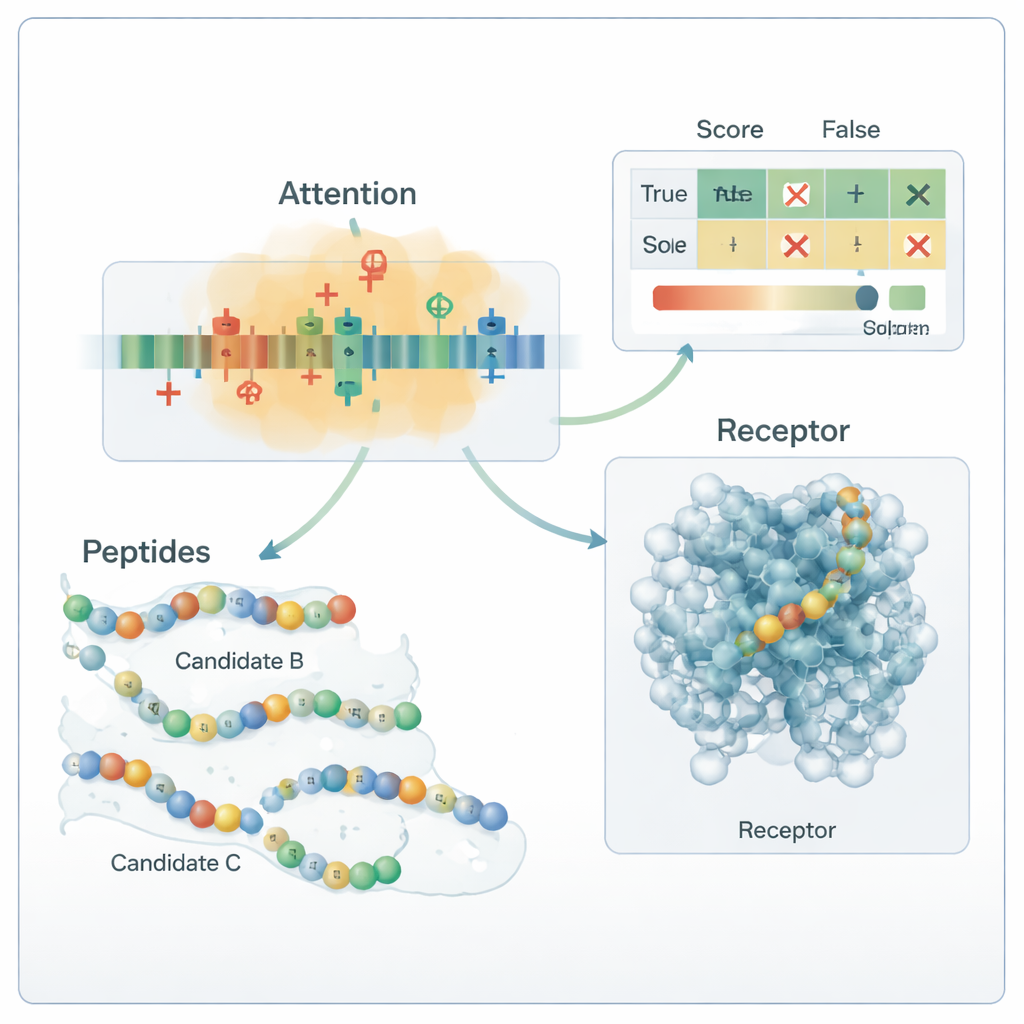
Der Kern der Pipeline ist ein hybrides Deep-Learning-Modell. Ein Convolutional Neural Network (CNN) scannt das Peptid entlang der Sequenz und sucht nach kurzen Motifen, die in aktiven Peptiden häufig vorkommen, ähnlich wie Filter in einem Bilderkennungssystem. Darüber hinaus lernt eine Attention‑Schicht, welche Positionen in der Sequenz am wichtigsten sind und fängt Fernbeziehungen zwischen weit entfernten Resten ein. Die Ausgabe dieser Sequenz‑Engine wird mit den globalen chemischen Deskriptoren verschmolzen und mehreren Standard‑Machine‑Learning‑Klassifikatoren übergeben – Support Vector Machines, Entscheidungsbäumen, k‑nächsten Nachbarn und Gradient‑Boosted Trees. Eine spezialisierte Optimierungsmethode namens OptimizedTPE passt automatisch die Parameter an und findet ein Gleichgewicht zwischen Genauigkeit und dem Risiko des Überanpassens. Der Attention‑Mechanismus liefert zudem restebezogene „Wichtigkeitskarten“, die Wissenschaftlerinnen und Wissenschaftlern zeigen, welche Teile jedes Peptids die Entscheidungen des Modells antreiben.
Neue Kandidaten erfinden und Datenleckage vermeiden
Um die geringe Anzahl bekannter antidiabetischer Peptide zu überwinden, fügte das Team eine Generierungsstufe hinzu, die nur in den Trainingsprozess einspeist. Sie nutzten eine Mischung aus Strategien – gesteuerte Mutationen, Motifrekombination und einen variationalen Autoencoder –, um neue Sequenzen vorzuschlagen, die bekannten aktiven Peptiden ähneln, sie aber nicht kopieren. Diese Kandidaten wurden dann durch strenge „Deskriptor‑Tore“ gesiebt, die realistische Ladung, Größe und Bindungsneigung erzwingen, sowie durch externe Werkzeuge, die die Ähnlichkeit zu bekannten bioaktiven Peptiden bewerten. Nur Sequenzen, die diese Filter passieren und deutlich unterscheidbar von allen Testpeptiden bleiben, werden als schwach gelabelte Positive ins Training übernommen; keine von ihnen wird jemals zur Bewertung des Modells genutzt. Dieser Ansatz erweiterte den Trainingssatz und bewahrte gleichzeitig eine saubere, unverzerrte Testbasis.
Wie gut das System funktioniert und was es bedeutet
Als das System mit einem völlig unabhängigen Panel von 180 experimentell untersuchten Peptiden aus jüngerer Literatur geprüft wurde, klassifizierte das Framework etwa 99 von 100 Sequenzen korrekt, mit Präzision und Recall nahe 0,99. Praktisch bedeutet das, dass es selten ein echtes antidiabetisches Peptid übersieht und selten ein inaktives Peptid fälschlich als vielversprechend einstuft. Analysen der Attention‑Karten und Mutationsprüfungen zeigten, dass das Modell chemisch sinnvolle Regeln gelernt hat: Es verlässt sich stark auf positiv geladene und bestimmte hydrophobe Reste, die für die Bindung an diabetesbezogene Ziele bekannt sind. Molekulare Docking‑Simulationen deuteten zudem darauf hin, dass einige der neu generierten Peptide plausible Kontakte mit dem menschlichen GLP‑1‑Rezeptor ausbilden können. Während diese Vorhersagen noch laborbestätigt werden müssen, zeigt die Studie einen reproduzierbaren, biologisch fundierten Weg, den riesigen Raum möglicher Peptidwirkstoffe zu erkunden und die wenigen zu priorisieren, die am wahrscheinlichsten zur Diabetesbehandlung beitragen könnten.
Zitation: Asl, Z.R., Rezaee, K., Ansari, M. et al. De novo generation and in silico screening of anti-diabetic peptide candidates via a deep learning–attention framework with physicochemical feature fusion. Sci Rep 16, 6580 (2026). https://doi.org/10.1038/s41598-026-39985-4
Schlüsselwörter: antidiabetische Peptide, Deep Learning, Wirkstoffforschung, Peptiddesign, GLP‑1-Rezeptor