Clear Sky Science · de
Interpretierbares maschinelles Lernen erklärt die Hemmung der Carboanhydrase durch konforme und kontrafaktische Vorhersage
Warum intelligentere Krebsmedikamente wichtig sind
Krebsmedikamente wirken häufig wie grobe Werkzeuge: Zwar greifen sie Tumorzellen an, können aber auch gesundes Gewebe treffen und schwere Nebenwirkungen verursachen. Eine vielversprechende Möglichkeit, diese Wirkung zu schärfen, besteht darin, bestimmte Varianten eines Enzyms namens Carboanhydrase zu blockieren, das Tumoren hilft, in sauerstoffarmen Umgebungen zu überleben. Viele Varianten dieses Enzyms sehen sich jedoch sehr ähnlich, was es schwierig macht, Wirkstoffe zu entwickeln, die die „schädlichen“ Varianten in Tumoren treffen, ohne die „gute“ Variante im ganzen Körper zu stören. Diese Studie zeigt, wie interpretierbares maschinelles Lernen Forschern helfen kann, diese Herausforderung zu meistern und selektivere, sicherere Wirkstoffkandidaten zu entwerfen.
Das Problem, das falsche Ziel zu treffen
Die menschliche Carboanhydrase (hCA) kommt in vielen Formen oder Isoformen vor. Zwei davon, IX und XII, stehen im Zusammenhang mit dem Überleben von Krebszellen in sauerstoffarmen Tumoren; ihre Blockade könnte das Fortschreiten der Krankheit bremsen und die Therapie verbessern. Isoform II ist jedoch weit verbreitet in gesundem Gewebe und besitzt eine aktive Stelle, die IX und XII sehr ähnlich ist. Wirkstoffe, die an alle drei binden, können unerwünschte Probleme wie metabolische Azidose und Sehstörungen auslösen. Traditionelle Labor- und Computermethoden stoßen an ihre Grenzen, weil Enzyme große, komplexe Moleküle sind und die Zahl möglicher wirkstoffähnlicher Verbindungen astronomisch hoch ist. Alle diese Verbindungen exhaustiv zu testen, weder im Labor noch am Computer, ist schlicht nicht machbar.
Aufbau einer sauberen und vertrauenswürdigen Datenbasis
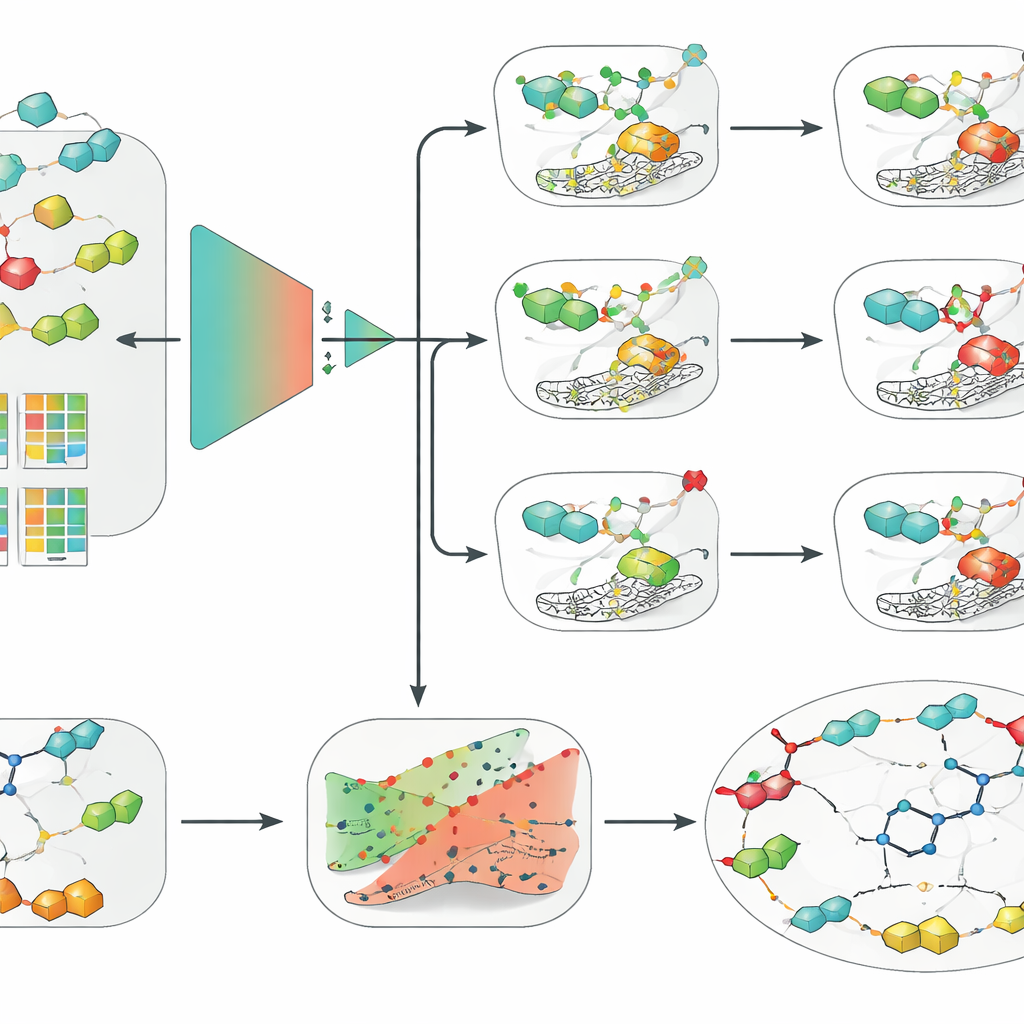
Die Autoren gingen dieses Problem an, indem sie zunächst eine sorgfältig bereinigte Datenbank aus Tausenden von Molekülen zusammenstellten, die gegen hCA II, IX und XII aus dem ChEMBL-Repository getestet worden waren. Sie standardisierten chemische Strukturen, entfernten zweifelhafte Messwerte und konzentrierten sich auf Verbindungen, die eine gemeinsame Zink-bindende Gruppe aufweisen, wie sie für diese Inhibitorenklasse typisch ist. Mit strengen Schwellenwerten kennzeichneten sie Moleküle als eindeutig aktiv oder eindeutig inaktiv und schieden unscharfe Fälle aus, die die Modelle verwirren könnten. Da deutlich mehr inaktive als aktive Moleküle vorlagen, balanceden sie die Daten, damit Lernalgorithmen nicht einfach die Mehrheitsklasse bevorzugen. Außerdem verwendeten sie eine „scaffold-basierte“ Aufteilung der Daten, sodass Trainings- und Testsätze unterschiedliche Kerngerüste enthielten; das liefert ein realistischeres Bild davon, wie gut die Modelle wirklich neue Verbindungen verarbeiten würden.
Einfache Modelle schlagen Deep Learning, wenn Daten knapp sind
Mit diesem kuratierten Datensatz verglich das Team eine breite Palette von Ansätzen, von klassischen Methoden des maschinellen Lernens wie logistischer Regression, Random Forests und Support-Vektor-Maschinen (SVMs) bis hin zu modernen tiefen neuronalen Netzen, einschließlich graphbasierter Modelle, die direkt auf molekularen Strukturen operieren. Sie kombinierten diese mit verschiedenen Möglichkeiten, Moleküle zu kodieren, etwa traditionellen handgemachten Deskriptoren, schlüsselbasierten Fingerprints und gelernten Einbettungen aus einem chemischen Sprachmodell. Über alle drei Enzymisoformen hinweg und unter der strengeren scaffold-basierten Evaluierung zeichnete sich eine Kombination durchgängig ab: eine SVM, gefüttert mit Extended-Connectivity-Fingerprints, einer strukturierten Beschreibung lokaler chemischer Umgebungen innerhalb eines Moleküls. Überraschenderweise übertraf dieses vergleichsweise simple Setup modischere graphbasierte und Deep-Learning-Modelle und unterstreicht, dass Datenqualität, sorgfältige Validierung und gute molekulare Deskriptoren wichtiger sein können als algorithmische Komplexität, wenn Datensätze moderater Größe vorliegen.
Zuverlässige Unsicherheiten und benutzerfreundliche Erklärungen
Die Forscher umhüllten ihr bestes SVM-Modell dann mit zwei zusätzlichen Schichten, die seine Vorhersagen in der praktischen Wirkstoffentwicklung nützlicher machen sollen. Zuerst setzten sie ein Framework namens konforme Vorhersage (conformal prediction) ein, das nicht nur eine einfache Ja-/Nein-Antwort liefert, sondern einen Bereich wahrscheinlicher Ergebnisse zusammen mit einer garantierten Fehlerrate angibt. Das erlaubt Wissenschaftlern, die Vorsicht des Modells zu justieren und Fälle zu erkennen, in denen das Modell wirklich unsicher ist. Zweitens verwendeten sie kontrafaktische Erklärungen, um die Entscheidungsgründe des Modells anschaulicher zu machen. Für ein gegebenes Molekül erzeugten sie eng verwandte Analoga, die das vorhergesagte Ergebnis von aktiv auf inaktiv oder umgekehrt kippen. Die Untersuchung dieser Paare für den klinischen Kandidaten SLC-0111, der IX und XII selektiv blockiert, nicht aber II, brachte eine wichtige Erkenntnis der medizinischen Chemie unabhängig wieder zutage: Kleine Veränderungen am „Schwanz“-Teil des Moleküls verändern stark, welche Isoform bevorzugt gebunden wird.
Von Algorithmen zu praktischen Werkzeugen für den Wirkstoffentwurf
Um ihren Ansatz zugänglich zu machen, packten die Autoren die drei SVM-Modelle, die Unsicherheitsschicht und die kontrafaktische Engine in ein grafisches Werkzeug namens CAInsight. Ein Nutzer kann eine textuelle Darstellung eines Moleküls eingeben und mit einem Klick die vorhergesagte Aktivität gegen hCA II, IX und XII erhalten, eine Abschätzung, wie vertrauenswürdig jede Vorhersage ist, sowie vorgeschlagene strukturelle Anpassungen, die die Aktivität erhöhen oder verringern könnten. Während die Modelle darauf ausgelegt sind, Moleküle als aktiv oder inaktiv zu klassifizieren statt in einem Schritt genaue Potenz- oder Selektivitätswerte zu liefern, reproduzieren sie bereits bekanntes Verhalten realer Wirkstoffkandidaten und unterscheiden subtile strukturelle Änderungen. Die Autoren weisen darauf hin, dass größere und einheitlichere Datensätze sowie eine gründlichere Analyse der Wahl von Aktivitätsschwellen die Leistung weiter verbessern könnten.
Was das für künftige Krebsmedikamente bedeutet
Einfach ausgedrückt zeigt diese Arbeit, dass sorgfältig gebaute und gut erklärte Modelle des maschinellen Lernens Chemikern dabei helfen können, Krebsmedikamente zu entwerfen, die besser zwischen ähnlich aussehenden Enzymzielen unterscheiden. Durch die Kombination robuster Statistik, Unsicherheitsabschätzungen und intuitiver „Was-wäre-wenn“-Beispiele sagt das Framework nicht nur voraus, welche Moleküle wahrscheinlich wirken, sondern liefert auch Hinweise, warum. Diese Art transparenter künstlicher Intelligenz könnte das virtuelle Screening beschleunigen, die generative Entwurfsarbeit für neue Verbindungen unterstützen und den Trial-and-Error-Aufwand im Labor reduzieren — letztlich die Entdeckung selektiverer und sichererer Behandlungen für Patienten fördern.
Zitation: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Schlüsselwörter: Carboanhydrase-Inhibitoren, interpretierbares maschinelles Lernen, Arzneimittelselektivität, konforme Vorhersage, kontrafaktische Erklärungen