Clear Sky Science · de
Kombinierte heterozygote CHAT-Genmutationen, eine Missense- und eine Spleißstellenvariante, bei zwei Geschwistern mit kongenitalem myasthenischem Syndrom
Wenn die Atmung ohne Vorwarnung versagt
Manche Kinder wirken bei der Geburt gesund und hören bei kleinen Fieberepisoden plötzlich auf zu atmen und brauchen eine Notbeatmung. Für ihre Familien sind diese Anfälle erschreckend und rätselhaft. Diese Studie untersucht zwei solche Geschwister aus Japan und führt ihre lebensbedrohlichen Schwäche‑ und Apnoeepisoden (Atemaussetzer) auf winzige Veränderungen in einem einzelnen Gen zurück, das die Kommunikation zwischen Nerven und Muskeln unterstützt. Anhand klinischer Hinweise, Gen-Sequenzierung und computerbasierter Proteinmodellierung zeigen die Forschenden, wie diese Mutationen wahrscheinlich ein Schlüsselenzym stören und Ärztinnen und Ärzten ein klareres Ziel für Diagnose und Behandlung geben.
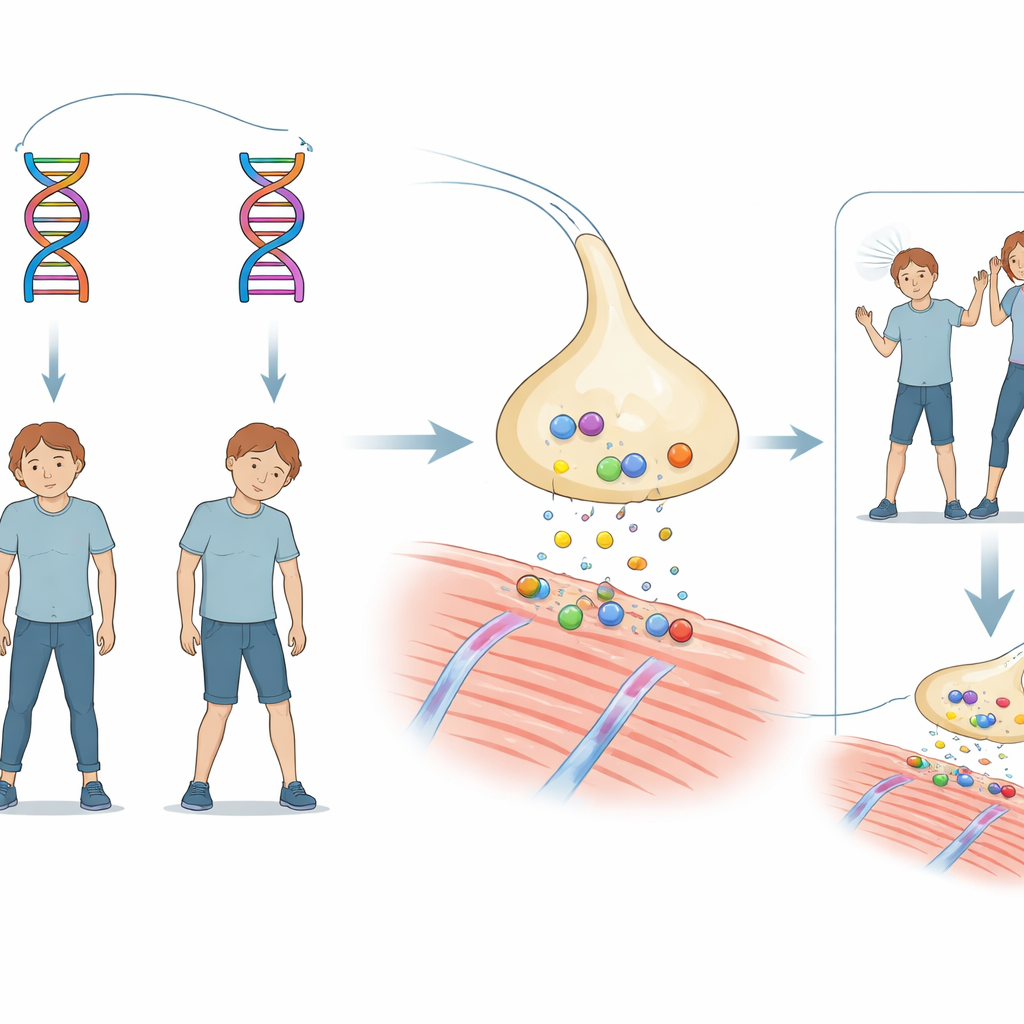
Ein familiäres Rätsel plötzlicher Schwäche
Die Geschichte dreht sich um einen Bruder und eine Schwester, die beide in der Säuglingszeit eine leicht verzögerte motorische Entwicklung zeigten. Im Alter von etwa 18 Monaten erlebten beide Anfälle von Apnoe und Bewusstseinsverlust bei Fiebern, die schwer genug waren, um eine Beatmung erforderlich zu machen. Mit dem Größerwerden litten beide weiterhin unter hängenden Augenlidern und generalisierter Muskelschwäche, ausgelöst durch Infektionen, Fieber oder Anstrengung. Die Gehirnscans waren normal, und häufige antikörpervermittelte Formen der Myasthenie (eine Erkrankung, bei der die Kommunikation zwischen Nerv und Muskel gestört ist) wurden ausgeschlossen. Ein Medikament, das das chemische Signal zwischen Nerven und Muskeln verstärkt, verbesserte jedoch eindeutig ihre Symptome, was auf eine seltene erbliche Erkrankung namens kongenitales myasthenisches Syndrom hinwies.
Die fehlerhaften Anweisungen finden
Um eine erbliche Ursache zu suchen, sequenzierten die Forschenden alle protein-kodierenden Gene der Geschwister und ihrer Eltern. Sie fanden, dass jedes Kind zwei unterschiedliche Veränderungen im selben Gen, CHAT, trug, das Cholinacetyltransferase kodiert—ein Enzym, das Acetylcholin herstellt, den Hauptbotenstoff, den Nerven verwenden, um Muskeln zu aktivieren. Eine Veränderung ersetzte einen einzelnen Baustein des Enzyms (eine Missense-Mutation bekannt als G411R). Die andere lag an einer kritischen Grenze, an der die Zelle normalerweise Genabschnitte beim RNA-Bildungsprozess schneidet und verbindet (eine Spleißstellenmutation bezeichnet als c.752+2T>C). Jeder Elternteil trug nur eine dieser Veränderungen und war gesund; nur die Kinder, die beide Veränderungen geerbt hatten, zeigten die Erkrankung, was darauf hindeutet, dass die Kombination der Mutationen die Enzymfunktion schwächt.
Untersuchen, wie ein versteckter Schnitt das Enzym verändert
Da die Forschenden nicht genügend natürliches CHAT-RNA aus Blutproben gewinnen konnten, verwendeten sie ein "Minigen-"Experiment. Sie klonierten den relevanten Genabschnitt in einen DNA-Vektor, führten entweder die normale oder die mutierte Version in kultivierte Zellen ein und untersuchten dann, wie die RNA verarbeitet wurde. Im normalen Konstrukt enthielt die RNA alle erwarteten Segmente. In der mutierten Version wurde ein ganzer Abschnitt, bekannt als Exon 5, übersprungen, doch der Gesamtleserahmen des Gens blieb intakt. Das bedeutete, dass das Enzym hergestellt würde, aber eine kurze innere Aminosäuresequenz fehlen würde. Evolutionsvergleiche zeigten, dass dieser fehlende Bereich zwischen Arten hoch konserviert ist, was darauf hindeutet, dass er eine wichtige strukturelle Rolle spielt.
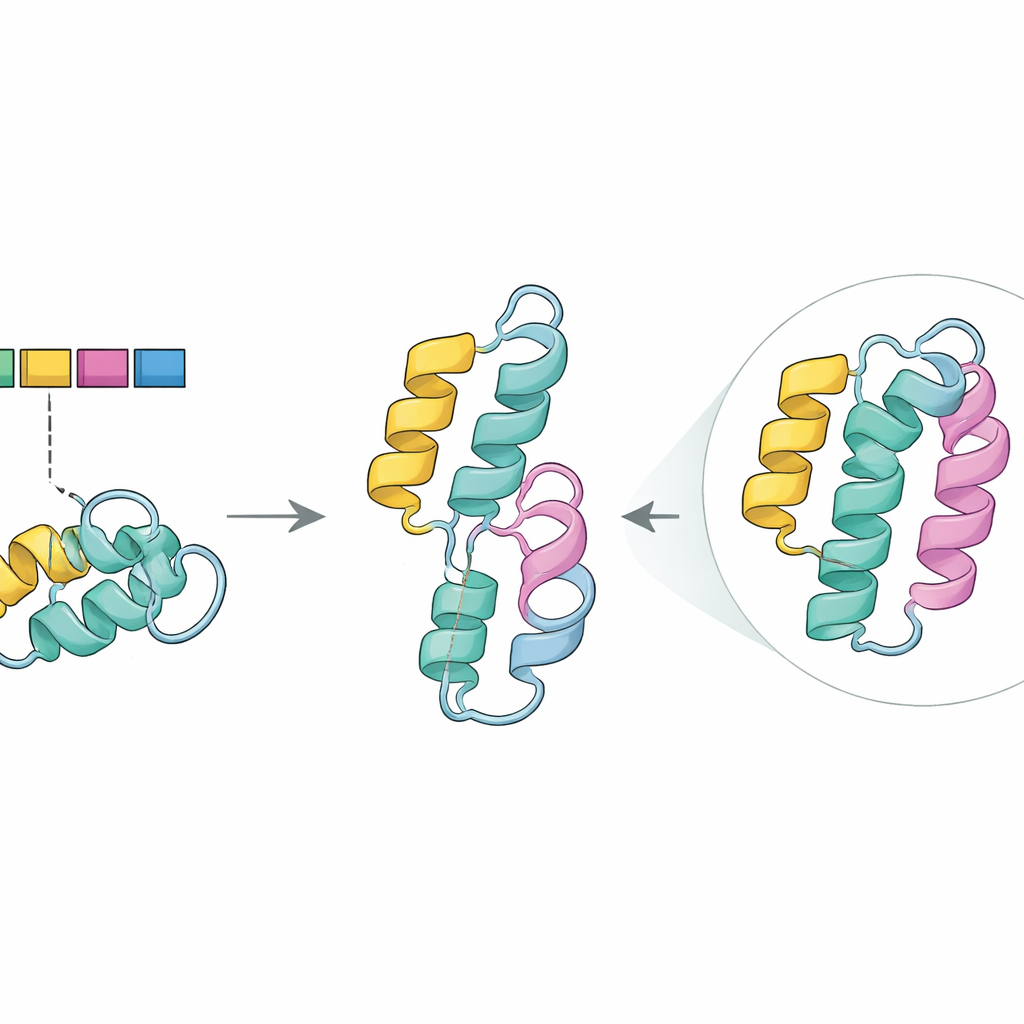
Strukturschäden in silico erkennen
Um diese Rolle zu untersuchen, wandte sich das Team AlphaFold2 zu, einem fortgeschrittenen Programm, das drei‑dimensionale Proteinformen aus Sequenzen vorhersagt. Im normalen Enzym bildet der von Exon 5 kodierte Abschnitt eine der dicht gepackten Spiralstrukturen (eine Alpha-Helix), die den Kern des Proteins stabilisieren. In der vorhergesagten mutierten Struktur verschwand diese Helix und hinterließ eine Lücke in einem Bereich, der aus früheren Arbeiten als entscheidend für die Aufrechterhaltung der Stabilität und für effiziente katalytische Vorgänge bekannt ist. Zusammen mit computergestützten Werkzeugen, die schädliche Mutationen kennzeichnen, stützen diese Ergebnisse die Idee, dass das Überspringen von Exon 5—insbesondere in Kombination mit der G411R-Veränderung auf der anderen Allelkopie—die Leistung des Enzyms beeinträchtigt, ohne es vollständig zu eliminieren—im Einklang mit den moderaten aber ernsten Symptomen der Geschwister.
Was das für Patientinnen, Patienten und Familien bedeutet
Die Studie kommt zu dem Schluss, dass die Kombination aus der G411R-Missense-Mutation und der neu identifizierten Spleißstellenmutation in CHAT sehr wahrscheinlich für das kongenitale myasthenische Syndrom der Geschwister verantwortlich ist. Indem die Forschenden durch den Minigene-Assay und die Strukturmodellierung zeigen, wie die Spleißstellenveränderung eine stabilisierende Helix aus dem Enzym entfernt, liefern sie eine mechanistische Erklärung, auf der Klinikerinnen, Kliniker und Forschende aufbauen können. Für betroffene Familien bietet eine solche Arbeit mehr als nur eine Diagnose: Sie unterstützt maßgeschneiderte Therapien mit Medikamenten, die die neuromuskuläre Signalübertragung verstärken, leitet die genetische Beratung für zukünftige Schwangerschaften und fügt dem Katalog hinzu, wie subtile Veränderungen in unserem genetischen Code die Muskelkraft und die grundlegende Funktion des Atmens tiefgreifend beeinflussen können.
Zitation: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Schlüsselwörter: kongenitales myasthenisches Syndrom, CHAT-Gen, Cholinacetyltransferase, Spleißstellenmutation, neuromuskuläre Endplatte