Clear Sky Science · de
Aufmerksamkeitsgesteuerte verbesserte Dekonvolution ermöglicht referenzfreie Zelltyp-Schätzung in der räumlichen Transkriptomik
Zellen im Kontext sehen
Die moderne Biologie kann die Aktivität von Tausenden Genen gleichzeitig lesen, nicht nur in isolierten Zellen, sondern direkt in dünnen Gewebsschnitten. Diese „räumliche Transkriptomik“-Perspektive zeigt, wo verschiedene Zellen leben und interagieren, doch jede Messung mischt oft Signale vieler benachbarter Zellen. Die Studie stellt eine neue rechnerische Methode vor, AGED genannt, die diese Mischungen entwirren und abschätzen kann, welche Zelltypen wo vorhanden sind — ohne ein separates, sorgfältig passendes Single-Cell-Referenzdatenset zu benötigen. 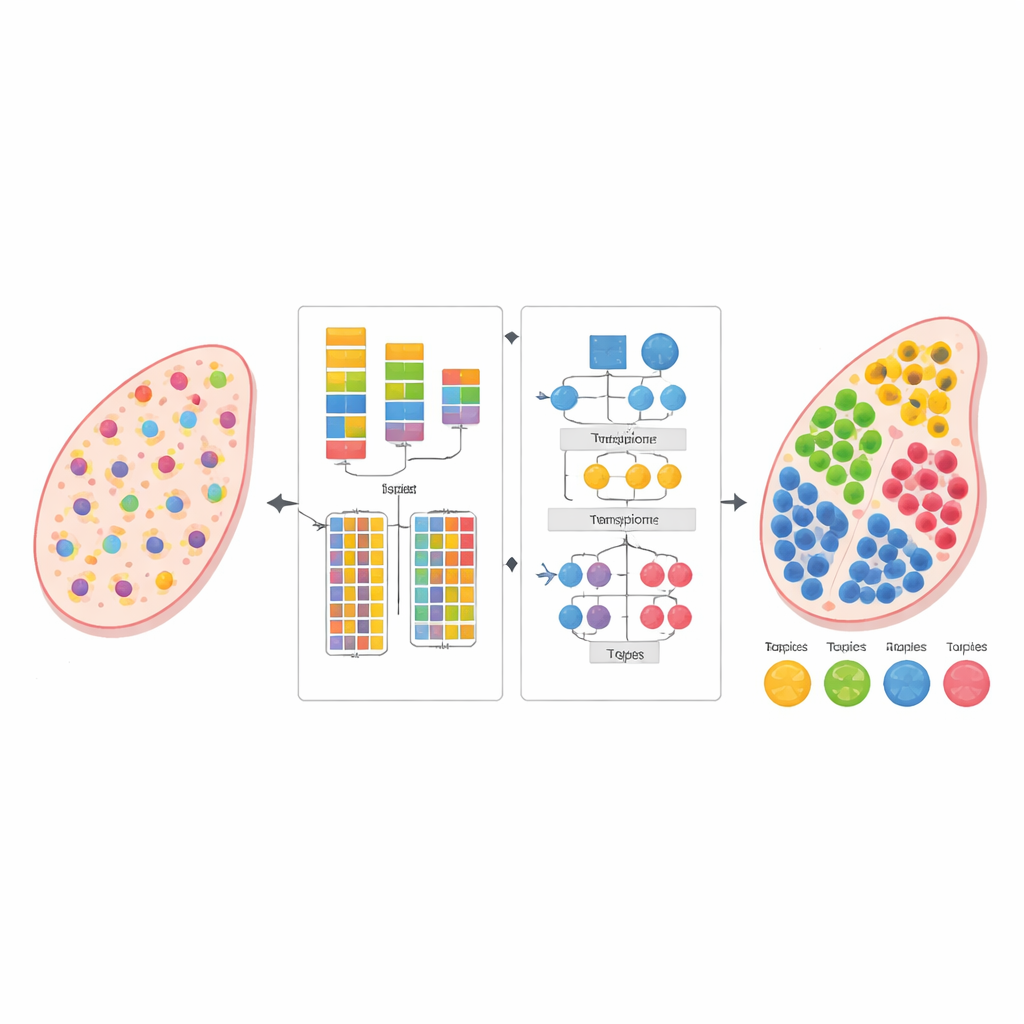
Warum das Kartieren von Zellen in Geweben schwierig ist
Räumliche Transkriptomik-Plattformen messen Genaktivität über ein Gitter von Spots auf einem Gewebsschnitt. Da die meisten Spots mehrere Zellen gleichzeitig erfassen, müssen Forschende die gemischten Signale mathematisch zerlegen, um die zugrundeliegenden Zelltypen und ihre Anteile zu rekonstruieren. Bestehende Werkzeuge stützen sich oft auf externe Single-Cell-Referenzatlanten desselben Gewebes. Solche Atlanten fehlen für seltene Gewebe, spezielle Krankheitszustände oder ungewöhnliche experimentelle Bedingungen, und selbst wenn sie vorliegen, passen sie möglicherweise nicht exakt, was Verzerrungen einführt. Referenzfreie Methoden vermeiden diese Abhängigkeit, tun sich jedoch schwer mit komplexen räumlichen Mustern, subtilen Genbeziehungen und der Herausforderung, vorher zu entscheiden, wie viele unterschiedliche Zelltypen überhaupt zu erwarten sind.
Eine zweistufige Strategie zum Entwirren von Mischungen
Die Autor*innen haben AGED als zweistufiges Rahmenwerk entworfen, das Ideen aus Statistik und modernem Deep Learning kombiniert. In der ersten Stufe testet die Methode eine Reihe von Möglichkeiten dafür, wie viele Zelltypen im Gewebe vorhanden sein könnten. Sie verwendet ein schnelles auf Aufmerksamkeit basierendes neuronales Netzwerk, bekannt als Performer, um Kandidaten-Dekompositionen zu lernen, und bewertet diese dann mit mehreren Kriterien gleichzeitig: wie gut das Modell die beobachteten Genzählungen rekonstruiert, wie deutlich sich die abgeleiteten Zellgruppen voneinander trennen und wie divers diese Gruppen sind. Ein Kurvenanpassungsverfahren findet einen „Knickpunkt“, an dem das Hinzufügen weiterer Zelltypen nur noch geringen Gewinn bringt, sodass die Methode automatisch eine geeignete Zahl auswählt, statt sich auf eine Benutzervorgabe zu verlassen.
Gelenkte Aufmerksamkeit zur Erfassung biologischer Strukturen
Sobald die Zahl der Zelltypen festgelegt ist, verfeinert AGED in der zweiten Stufe die Lösung mit einer reichhaltigeren auf Aufmerksamkeit basierenden Architektur. Sie beginnt mit einem statistischen Topic-Modell, das jeden Gewebsspot als Mischung aus verborgenen „Themen“ behandelt — hier stellvertretend für Zelltypen — und jeden Zelltyp als charakteristisches Genmuster. Diese initialen Themen liefern globale Struktur. Das Modell schichtet dann mehrere Aufmerksamkeitsmechanismen darüber: einer verbindet die statistischen Themen mit dem neuronalen Netzwerk, ein anderer bündelt Informationen aus benachbarten Spots im physischen Raum, und ein dritter verknüpft Themen direkt mit Genen. Ein Gate-System lässt das Modell für jeden Einzelfall entscheiden, wie sehr es den vorherigen statistischen Mustern im Vergleich zu den lokalen Daten vertrauen soll. Zusätzliche Zwänge fördern sparsamer Lösungen, die der biologischen Realität entsprechen, dass die meisten Gewebsorte von nur wenigen dominanten Zelltypen geprägt sind. 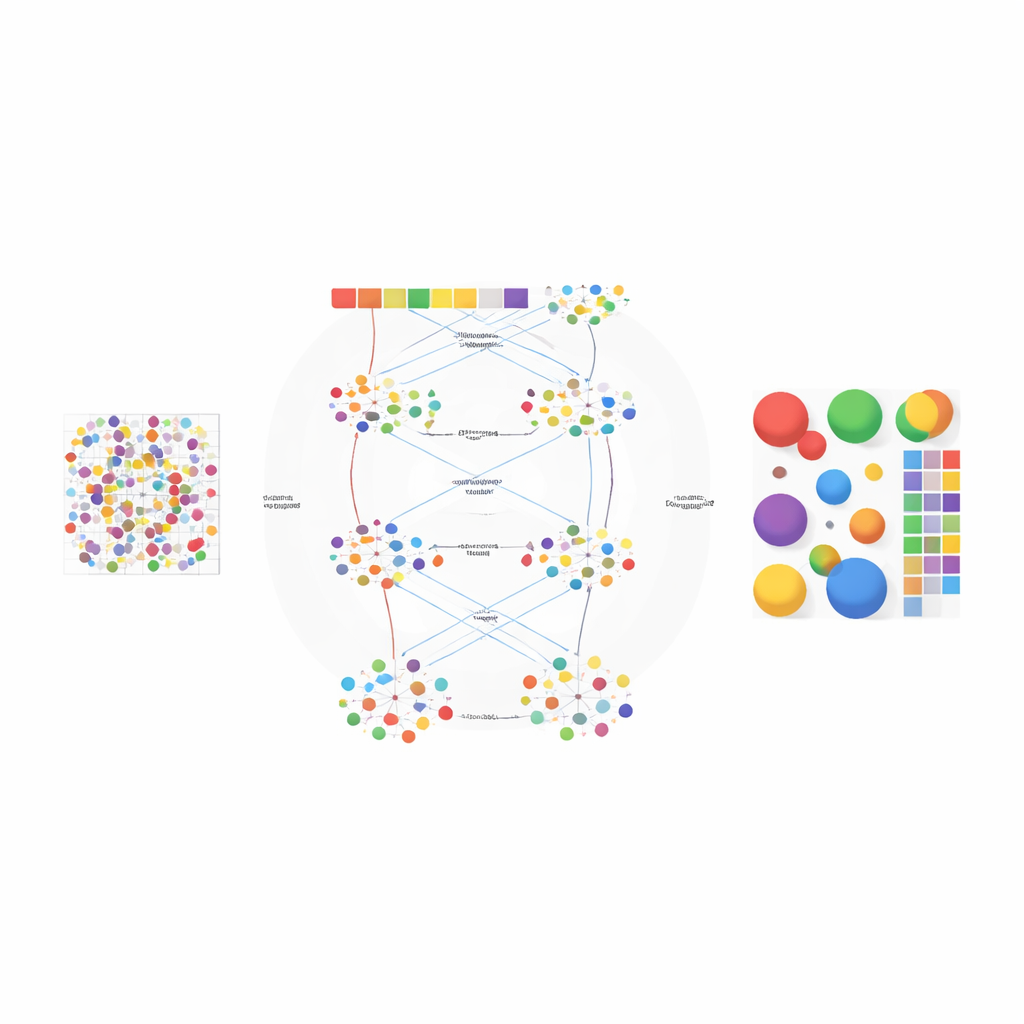
Wie die Methode getestet wurde
Die Forschenden evaluierten AGED an mehreren Datentypen. In simuliertem Maus-Riechkolbengewebe rekonstruierte die Methode vier bekannte anatomische Schichten und stimmte den wahren Zellzusammensetzungen näher überein als weit verbreitete referenzbasierte und referenzfreie Werkzeuge, wobei sie sowohl hohe Korrelation mit der Ground Truth als auch geringe Rekonstruktionsfehler erreichte. Im menschlichen duktalen Adenokarzinom der Bauchspeicheldrüse wählte AGED automatisch eine 20-Zelltypen-Lösung, die mit von Patholog*innen annotierten Regionen wie Tumor, Gang und normaler Bauchspeicheldrüse übereinstimmte und andere Methoden bei einem strukturellen Ähnlichkeitsmaß, das inferierte Karten mit der sichtbaren Gewebestruktur vergleicht, übertraf. Im menschlichen Thymus trennte AGED wichtige Zellpopulationen genau und erfasste eine biologisch erwartete negative Beziehung zwischen zwei spezialisierten epithelialen Zelltypen — ein Muster, das konkurrierende Ansätze nicht reproduzieren konnten. Weitere Analysen an zusätzlichen Datensätzen und in nahezu Einzelzellauflösung untermauerten die Robustheit der Methode.
Was das für die Zukunft bedeutet
Für Nicht-Spezialisten kann AGED als ein intelligenter Entmischungs-Mechanismus für komplexe Gewebe betrachtet werden: Es lernt, wie viele unterschiedliche Zellgemeinschaften vorhanden sind, wo sie liegen und welche Gene sie definieren — alles aus den räumlichen Daten selbst. Indem interpretierbare statistische Modelle mit flexiblen auf Aufmerksamkeit basierenden neuronalen Netzen verknüpft werden, bietet das Framework sowohl Genauigkeit als auch Einsicht, selbst wenn kein passender Referenzatlas existiert. Das macht es zu einem praktischen Werkzeug zur Erforschung der Gewebeorganisation in Gesundheit und Krankheit, von Hirnschichten über Tumoren bis zu Immunorganen, und weist auf eine breitere Strategie hin, wie Vorwissen genutzt werden kann, um leistungsfähige, aber oft undurchsichtige maschinelle Lernmodelle in der Biologie zu leiten.
Zitation: Yang, X., Wang, Y. & Chen, X. Attention-guided enhanced deconvolution enables reference-free cell type estimation in spatial transcriptomics. Sci Rep 16, 8097 (2026). https://doi.org/10.1038/s41598-026-39703-0
Schlüsselwörter: räumliche Transkriptomik, Zelltyp-Dekonvolution, Deep Learning, Gewebearchitektur, referenzfreie Analyse