Clear Sky Science · de
Gengetriebener analytischer Lernansatz für eine präzise Brustkrebsdiagnose
Warum diese Forschung für Patientinnen und Familien wichtig ist
Brustkrebs ist heute die weltweit am häufigsten diagnostizierte Krebserkrankung bei Frauen, und Patientinnen, die auf dem Papier dieselbe Erkrankung zu haben scheinen, können sehr unterschiedliche Verläufe aufweisen. Diese Studie zeigt, wie Muster in Tausenden von Genen kombiniert mit einem sorgfältig entwickelten System der künstlichen Intelligenz Ärzten dabei helfen können, verlässlicher zu unterscheiden, wer Krebs hat und wie schwerwiegend dieser sein könnte – und das allein auf Basis realer Patientendaten und einer kompakten Auswahl entscheidender Gene.
Von vielen Risikofaktoren zur Sprache der Gene
Das Risiko für Brustkrebs wird von vielen Einflüssen geformt: vererbte Genveränderungen, Hormone, Körpergewicht, Lebensstil und mehr. Sobald Krebs entsteht, wird sein Verhalten dadurch bestimmt, welche Gene in jedem Tumor ein- oder ausgeschaltet sind. Moderne Sequenzierverfahren können die Aktivität von Zehntausenden Genen gleichzeitig messen, doch aus diesem Ozean an Zahlen klare Ja‑/Nein‑Antworten für Diagnose und Prognose abzuleiten, ist schwierig. Traditionelle Rechenmethoden betrachten Gene oft eins nach dem anderen und können dabei übersehen, wie Gengruppen zusammenwirken, oder sie liefern nur auf einem Datensatz gute Ergebnisse und versagen bei anderen.
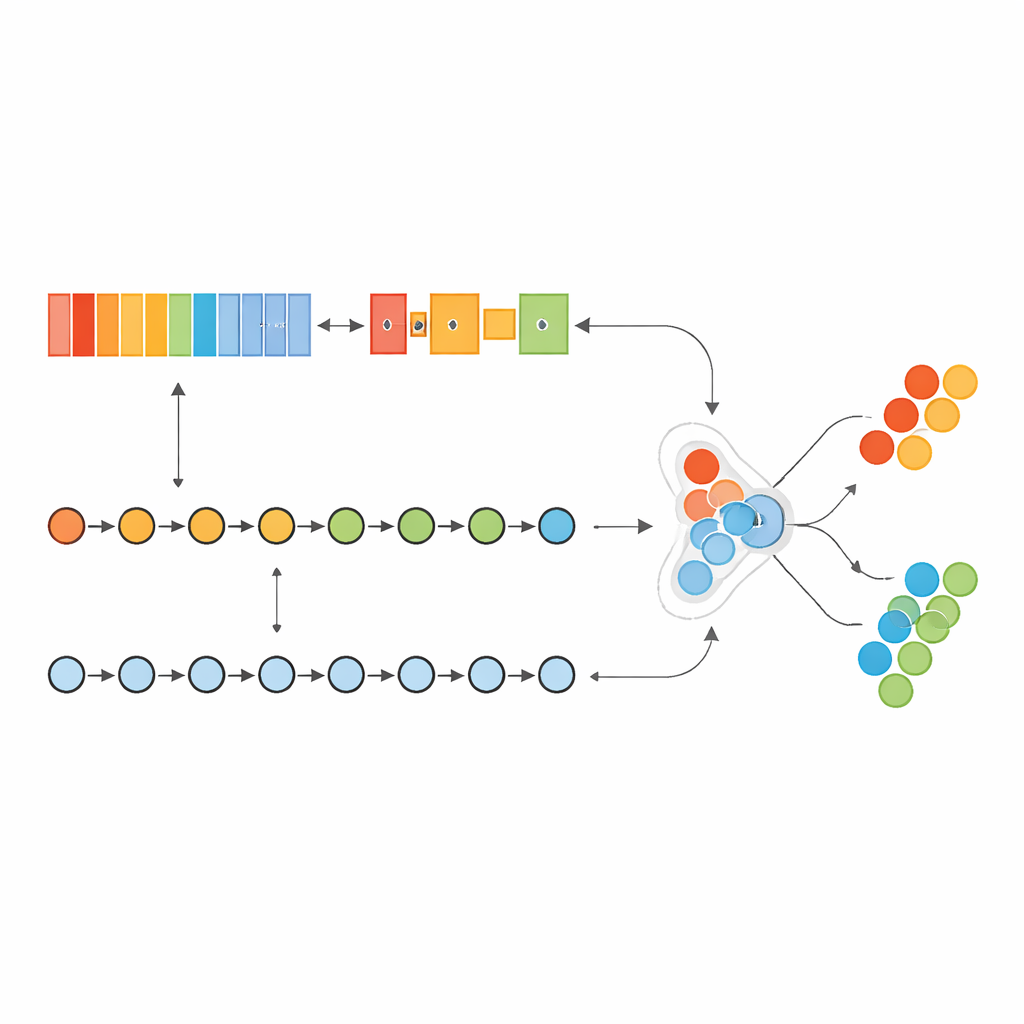
Einem Zweifach‑Modell beibringen, Genmuster zu lesen
Die Autoren entwickelten ein „hybrides“ Deep‑Learning‑Modell, das ein wenig wie zwei spezialisierte Gehirne zusammenarbeitet. Ein Teil, inspiriert von der Bildanalyse, scannt eine geordnete Liste von Genen, um lokale Muster zu erkennen – Gruppen von Genen, deren gemeinsame Aktivität auf Krebs hinweist. Der andere Teil behandelt dieselben Gene als Sequenz und lernt, wie frühe „Driver“-Gene und spätere „Downstream“-Gene einander entlang der Liste beeinflussen. Durch die Kombination dieser beiden Perspektiven kann das Modell sowohl kurzreichweitige als auch langreichweitige Beziehungen innerhalb des genetischen Fingerabdrucks des Tumors erfassen.
Finden eines stabilen Kernsets signalgebender Gene
Anstatt alle 17.815 gemessenen Gene in das Modell einzuspeisen, entwarf das Team eine strikte, „leakage‑freie“ Pipeline, um nur die informativsten Gene auszuwählen. Mit einem standardisierten Korrelationsmaß innerhalb wiederholter Cross‑Checking‑Schleifen bewerteten sie die Gene wiederholt danach, wie stark deren Aktivität mit dem Krebsstatus korrelierte. Anschließend behielten sie nur jene Gene bei, die in allen Trainingsaufteilungen konsequent an der Spitze auftauchten, was zu einer stabilen Signatur von 236 Genen führte. Die Forschenden kartierten außerdem, wie diese Gene miteinander interagieren, und zeigten, dass viele enge Netzwerke bilden, die mit Tumorwachstum, Stoffwechsel, Immunantwort und dem umgebenden Gewebe zusammenhängen – ein Hinweis darauf, dass die ausgewählte Gruppe reale biologische Vorgänge widerspiegelt und kein zufälliges Rauschen ist.
Das Modell auf die Probe stellen
Das hybride System wurde an Brustkrebsproben aus dem Cancer Genome Atlas trainiert und optimiert und anschließend mit einem vollständig separaten Datensatz, bekannt als METABRIC, geprüft. Um mit der Tatsache umzugehen, dass Krebsproben die Normalproben deutlich überwiegen, erzeugten die Autoren keine künstlichen Daten; stattdessen passten sie an, wie stark das Modell Fehler bei der selteneren Klasse „gewichtet“. Nach einer automatisierten Suche nach den besten Einstellungen erreichte das Modell auf dem Hauptdatensatz nahezu perfekte Werte, erkannte fast alle Krebsfälle korrekt und produzierte praktisch keine Fehlalarme. Wichtig ist, dass die Leistung auch beim Einsatz auf der externen METABRIC‑Kohorte extrem hoch und sehr stabil blieb, was darauf hindeutet, dass der Ansatz über eine einzelne Studie oder Klinik hinaus generalisieren kann.
Was das für die zukünftige Versorgung bedeutet
Einfach ausgedrückt liefert diese Arbeit eine fein abgestimmte, zweiteilige KI, die einen kompakten 236‑Gen‑Code liest, um mit bemerkenswerter Genauigkeit und Zuverlässigkeit zwischen krebsartigen und nicht‑krebsartigen Brustproben zu unterscheiden – selbst unter lauten (störenden) Bedingungen. Während die aktuelle Studie sich nur auf Genaktivität und retrospektive Patientendaten stützt, ebnen ihre Methoden den Weg für künftige Werkzeuge, die mehrere Datentypen kombinieren könnten – etwa Gewebebilder und zusätzliche molekulare Schichten – und klare Erklärungen liefern, welche Gene jede Vorhersage antreiben. Mit weiterer Validierung in prospektiven klinischen Studien könnte ein solches System zur universellen Grundlage für eine präzisionsmedizinische Brustkrebsdiagnose werden und Ärztinnen und Ärzten helfen, die Behandlung anhand der genetischen „Signatur“ des Tumors jeder Patientin zu individualisieren.
Zitation: Hesham, F., Abbassy, M.M. & Abdalla, M. Gene driven analytical learning model for accurate breast cancer diagnosis. Sci Rep 16, 8155 (2026). https://doi.org/10.1038/s41598-026-39430-6
Schlüsselwörter: Brustkrebsdiagnose, Genexpression, Tiefes Lernen, CNN-BiLSTM, Präzisionsonkologie