Clear Sky Science · de
FAM120A – ein Protein, das in das ALS‑Krankheitsnetzwerk eingebunden ist
Warum das für Betroffene und ihre Familien wichtig ist
Amyotrophe Lateralsklerose (ALS) ist eine verheerende Erkrankung, die Menschen allmählich lähmt, indem die Nervenzellen, die Bewegungen steuern, absterben. Bis heute verstehen wir noch nicht vollständig, warum diese Motoneurone zugrunde gehen, und wirksame Behandlungen sind rar. Diese Studie richtet den Blick auf ein wenig bekanntes Protein namens FAM120A und legt nahe, dass es Nervenzellen helfen könnte, mit Stress umzugehen und die Ansammlung schädlicher Proteinklumpen zu verhindern – ein Kennzeichen von ALS. Indem sie offenlegt, wie sich dieses Protein während der Erkrankung verhält, eröffnet die Arbeit einen neuen Weg zum Verständnis und möglicherweise irgendwann zur Behandlung von ALS.
Ein versteckter Akteur in einem dicht gedrängten Gennetzwerk
Die Forschenden begannen nicht am Labortisch, sondern am Computer. Sie nutzten einen „konvergenten Analyse“-Ansatz, um zahlreiche bestehende Datensätze über mit ALS verknüpfte Gene und deren Interaktionen zu kombinieren. Diese netzwerkartige Sicht erlaubte es ihnen, Cluster von Proteinen zu erkennen, die in zentralen zellulären Prozessen zusammenarbeiten, insbesondere in solchen, die mit RNA‑Handhabung und Protein‑Qualitätskontrolle zu tun haben – beides bekannte Problemfelder bei ALS. Innerhalb eines solchen Clusters trat FAM120A als zuvor übersehener, aber hoch vernetzter Faktor hervor, der mit mehreren etablierten ALS‑bezogenen Proteinen interagiert. Seine bekannten Rollen bei der Unterstützung von Zellen gegen oxidativen Stress und bei der RNA‑Regulation machten es zu einem starken Kandidaten für weitere Untersuchungen.
Ein verletzliches Protein im Verlauf der Erkrankung verfolgen
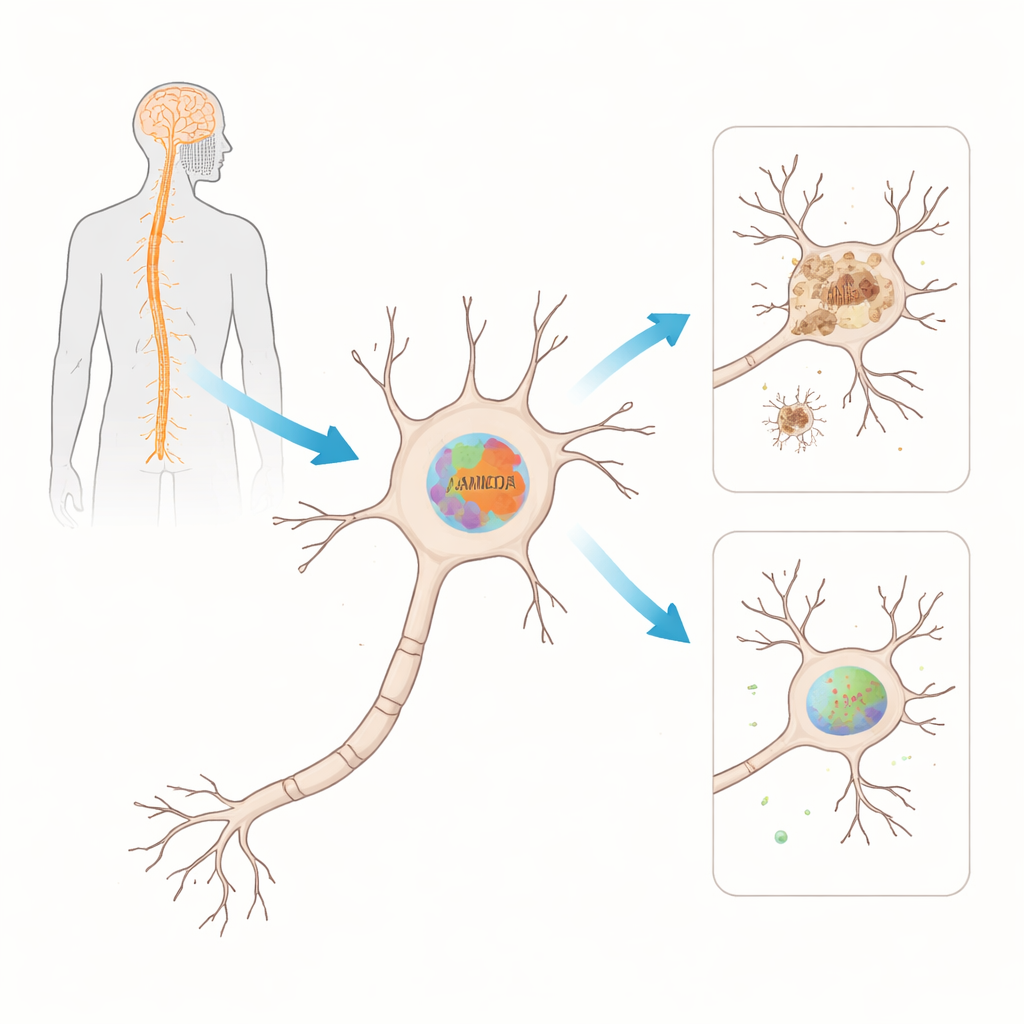
Um zu prüfen, ob FAM120A in der Tat eine Rolle bei ALS spielt, nutzte das Team ein weit verbreitetes Mausmodell, das eine mutierte Version des SOD1‑Gens trägt – eines der ersten genetisch identifizierten Ursachen von ALS. Sie bestimmten sowohl die RNA‑Botschaften als auch die Proteinspiegel der Mausversion Fam120A im Rückenmark über die Zeit, von vor Auftreten der Symptome bis zur späten Erkrankung. Frühzeitig sanken die RNA‑Spiegel von Fam120A im Rückenmark, bevor die Tiere klare Krankheitsanzeichen zeigten. Später, als die Lähmung sich entwickelte, war das Fam120A‑Protein selbst deutlich vermindert. Diese Asynchronie – zuerst Änderungen auf RNA‑Ebenen, später auf Proteinebene – deutet darauf hin, dass mehrere Regulierungsebenen zusammenbrechen, während die Erkrankung fortschreitet.
Wo im Rückenmark dieses Protein vorkommt
Als Nächstes fragten die Wissenschaftler, wo genau Fam120A im Rückenmark lokalisiert ist. Mit fluoreszenzmikroskopischen Aufnahmen von Gewebeschnitten beobachteten sie, dass Fam120A vorwiegend in Neuronen des ventralen Horns vorkommt – dem Bereich, der reich an Motoneuronen ist und bei ALS degeneriert. In Tieren im späten Krankheitsstadium sahen sie etwas Signal in Stützzellen, den Astrozyten, doch das dominante Muster blieb neuronal. Diese Beobachtungen verknüpfen Fam120A direkt mit den Zellen, die bei ALS ausfallen, und stützen die Idee, dass dessen Verlust ihre Fähigkeit, zellulären Stress zu bewältigen, schwächen und so zum Abbau der motorischen Funktion beitragen könnte.
Mehr FAM120A in nervenähnlichen Zellen einsetzen
Das Team ging dann zu kultivierten nervenähnlichen Zellen über, um zu erforschen, was FAM120A tatsächlich bewirkt. Sie konstruierten diese Zellen so, dass sie entweder normales oder mutiertes SOD1 produzierten, welches dazu neigt, toxische Aggregate zu bilden, und zwangen die Zellen zusätzlich dazu, vermehrt menschliches FAM120A herzustellen. In Anwesenheit des mutierten SOD1 verringerte die Erhöhung von FAM120A sowohl die Menge an unlöslichem SOD1, die biochemisch nachgewiesen wurde, als auch die Anzahl sichtbarer Aggregate im Mikroskop deutlich. Wichtig ist, dass FAM120A wenig Einfluss auf die normale Form von SOD1 hatte, was darauf hindeutet, dass es möglicherweise speziell Zellen dabei hilft, fehlgefaltete oder aggregationsneigende Proteine zu kontrollieren – ein zentrales Problem bei ALS und anderen neurodegenerativen Erkrankungen.
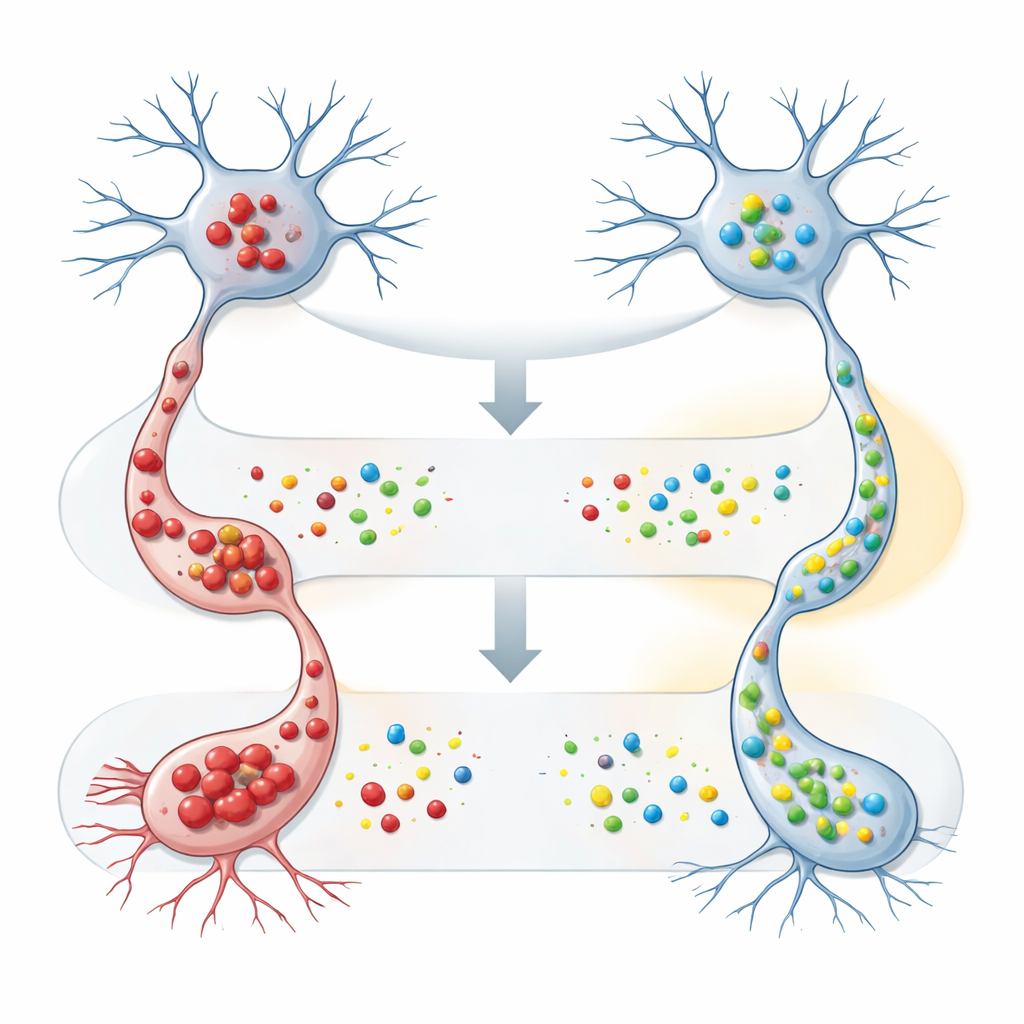
Eine breitere Karte molekularer Verbündeter und Gegenspieler erstellen
Über diese Experimente hinaus untersuchten die Forschenden das erweiterte Interaktionsnetzwerk von FAM120A. Sie bestätigten, dass es physisch mit PURA assoziiert, einem RNA‑bindenden Protein, das bereits mit Gehirnentwicklung und Neurodegeneration in Verbindung gebracht wurde, und stellten fest, dass auch die PURA‑Spiegel im Rückenmark der ALS‑Maus fallen, wenn auch später im Krankheitsverlauf. Sie heben weitere Verbindungen zwischen FAM120A, seinem Antisense‑Partnergen FAM120Aos und einem weiteren RNA‑bindenden Protein, ELAVL1, hervor, das Entzündungs‑ und Stressantwortgene im Gehirn reguliert. Dieses wachsende Netz von Verknüpfungen platziert FAM120A an der Schnittstelle von RNA‑Regulation, Stressantworten und Protein‑Qualitätskontrolle – genau den Systemen, die bei ALS versagen.
Was das für zukünftige ALS‑Behandlungen bedeuten könnte
Zusammengefasst deuten die Ergebnisse darauf hin, dass FAM120A nicht nur ein Beobachter ist, sondern ein bedeutsamer Teil des ALS‑Krankheitsnetzwerks. Sein früher Rückgang in besonders verletzlichen Motoneuronen, seine physischen Verknüpfungen zu anderen RNA‑regulierenden Proteinen und seine Fähigkeit, toxische SOD1‑Klumpen in Zellen zu reduzieren, sprechen für eine schützende Rolle bei der Aufrechterhaltung des Proteingleichgewichts. Zwar bleibt noch viel Arbeit zu tun – insbesondere um zu klären, ob ähnliche Veränderungen bei Menschen mit ALS und in anderen Krankheitsmodellen auftreten – doch rückt FAM120A als vielversprechendes Ziel für weitere Studien und möglicherweise künftige Therapien, die die Gesundheit der Motoneurone bewahren sollen, in den Fokus.
Zitation: Vicencio, E., Gomez, L., Beltran, S. et al. FAM120A - a protein inserted in the ALS disease network. Sci Rep 16, 8200 (2026). https://doi.org/10.1038/s41598-026-39329-2
Schlüsselwörter: amyotrophe Lateralsklerose, Motoneurone, Proteinaggregation, RNA‑bindende Proteine, Neurodegeneration