Clear Sky Science · de
Transkriptionelle Aktivierung von PPP1R14C durch KLF7 entfesselt CDK1-Aktivität und fördert das Plattenepithelkarzinom der Lunge
Warum diese Entdeckung beim Lungenkrebs wichtig ist
Lungenkrebs bleibt weltweit die häufigste krebsbedingte Todesursache, und eine Hauptform — das Plattenepithelkarzinom der Lunge — ist im Zeitalter gezielter Medikamente zurückgeblieben. Anders als einige andere Lungen tumoren, die sich mit Wirkstoffen gegen bestimmte Mutationen behandeln lassen, müssen Ärzte bei diesem Subtyp häufig auf Chemotherapie und Immuntherapie zurückgreifen, die nicht für alle Patienten funktionieren. Diese Studie enthüllt einen zuvor verborgenen Steuerkreislauf in Plattenepithelkrebszellen der Lunge, der wie das Lösen der Bremse der Zellteilung wirkt, und zeigt eine konkrete Schwachstelle, die künftige Medikamente ausnutzen könnten.
Eine fehlende Verbindung bei einem schwer zu behandelnden Lungenkrebs
Die Forschenden begannen damit, große öffentliche Krebsdatenbanken zu durchsuchen, um zu prüfen, ob bestimmte Gene im Plattenepithelkarzinom der Lunge auffielen. Eines mit dem Namen PPP1R14C trat in Tumorproben im Vergleich zu normalem Lungengewebe durchgehend in hohen Mengen auf. Seine Menge stieg, je weiter die Tumoren fortschritten, und Patientinnen und Patienten, deren Tumore mehr dieses Moleküls produzierten, lebten tendenziell kürzer. Diese Muster zeigten sich sowohl auf RNA-Ebene — den Nachrichten, die Zellen zur Proteinproduktion nutzen — als auch auf Proteinebene, was darauf hindeutet, dass PPP1R14C nicht nur präsent, sondern aktiv an der Krankheitsförderung beteiligt ist. 
Wie Lungentumoren die Bremse lösen
Um zu verstehen, warum PPP1R14C in diesen Tumoren so reichlich vorkommt, richtete das Team den Blick auf den Ein-/Ausschalter des Gens, dessen Promotor. Durch die Kombination mehrerer Datenbanken, die nachverfolgen, wo verschiedene Kontrollproteine an DNA binden, identifizierten sie einen Faktor namens KLF7 als Hauptverdächtigen. In im Labor gezüchteten Plattenepithelkrebszellen der Lunge führte eine Erhöhung von KLF7 zu einem Anstieg von PPP1R14C, während eine Herunterregulierung von KLF7 dessen Menge stark verringerte. Experimente, in denen der PPP1R14C-Promotor an einen lichtemittierenden Reporter gekoppelt wurde, bestätigten, dass KLF7 diesen Schalter direkt umlegen kann; die Veränderung einer kurzen DNA-Sequenz, an der KLF7 andockt, beseitigte den Effekt. Eine Technik, die DNA, die an KLF7 gebunden ist, aus intakten Zellen zieht, zeigte, dass dieser Faktor physisch am PPP1R14C-Promotor sitzt, womit bewiesen ist, dass KLF7 dieses Gen direkt einschaltet.
Vom genetischen Schalter zum aggressiven Verhalten
Sobald klar war, was PPP1R14C antreibt, untersuchten die Wissenschaftler, was das Molekül tatsächlich bewirkt. In Zelllinien des Plattenepithelkarzinoms der Lunge senkten sie PPP1R14C mittels gentechnischer Werkzeuge und beobachteten das Verhalten der Zellen. Zellen ohne PPP1R14C wuchsen langsamer, bildeten weniger Kolonien, durchdrangen eine Gelbarriere seltener und neigten eher zu programmierter Zellbindung (Apoptose). Das spiegelbildliche Ergebnis war ebenfalls zu sehen: Zellen, die künstlich mehr PPP1R14C produzierten, teilten sich schneller, bildeten mehr Kolonien und invasiveres Verhalten. Wurden diese veränderten Zellen in Mäuse implantiert, wuchsen Tumore mit reduziertem PPP1R14C kleiner und wogen weniger. Zusammengenommen zeigen diese Befunde, dass PPP1R14C kein Nebenakteur ist, sondern aktiv krebsfördernde Eigenschaften antreibt.
Ein schrittweiser Blick in den Motor des Zellzyklus
Vertiefende Untersuchungen betrachteten, welche zellulären Programme von PPP1R14C abhängen. Breite Analysen der Genaktivität zeigten, dass das Entfernen von PPP1R14C besonders Gene stört, die die kritische G2/M-Kontrollstelle regulieren — den Punkt, an dem sich eine Zelle zur Teilung verpflichtet. Im Zentrum dieser Kontrollstelle steht CDK1, ein Schlüsselschalter für den Eintritt in die Mitose. In Krebszellen mit hohem PPP1R14C trug CDK1 eine aktivierende Phosphatmarke und seine nachgeschalteten Ziele waren aktiviert, was ein grünes Licht für die Teilung signalisierte. Wurde PPP1R14C reduziert, verschwand diese Aktivierung. Biochemische Experimente zeigten den Grund: PPP1R14C bindet an ein zelluläres „Radiergummi“-Enzym namens PP1, das normalerweise die aktivierende Marke von CDK1 entfernt. Indem PPP1R14C PP1 blockiert, verhindert es dessen Zugriff auf CDK1, sodass das Aktivierungssignal bestehen bleibt und die Zellen weiter zyklieren. 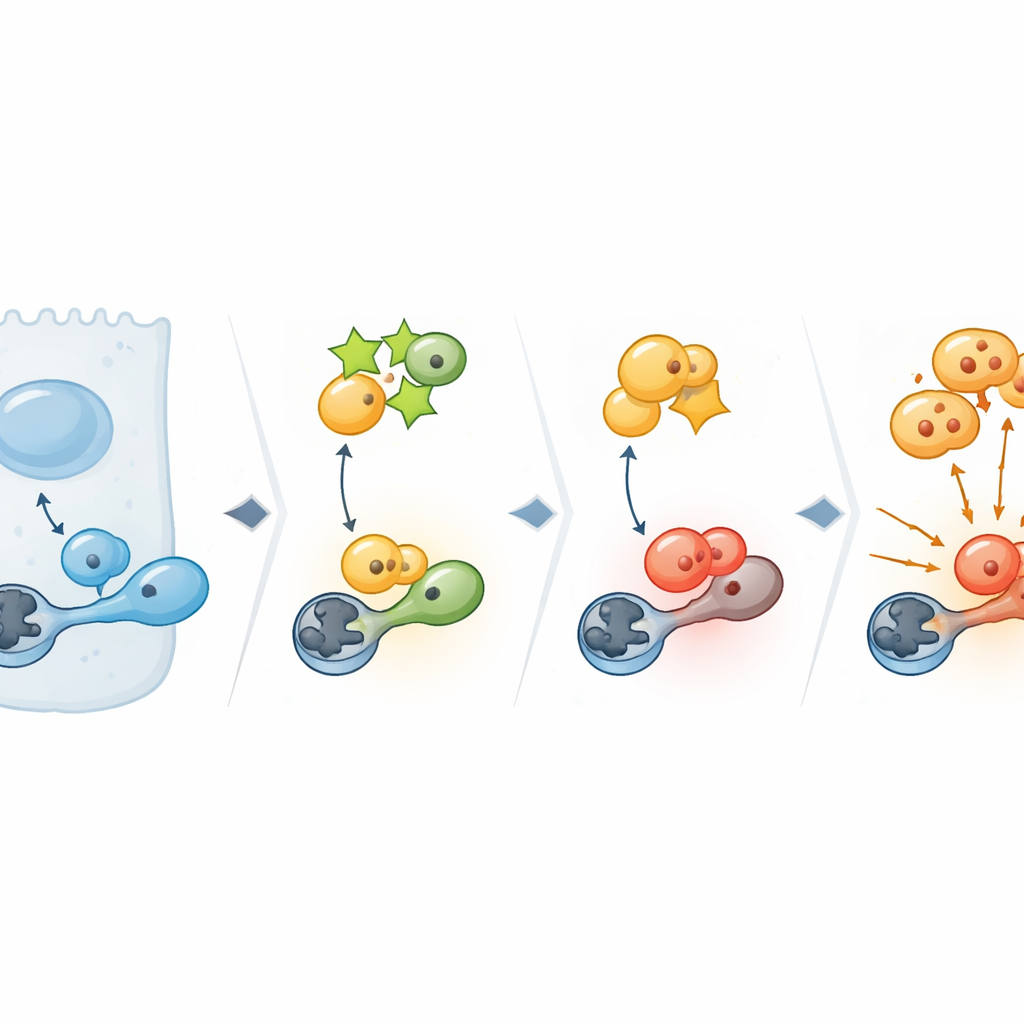
Aus einer molekularen Erkenntnis eine Behandlungsidee formen
Der vielversprechendste Teil der Arbeit war der Test eines Wirkstoffs, der CDK1 direkt blockiert. In Zellen mit Übermaß an PPP1R14C beseitigte dieser CDK1-Inhibitor den Wachstums-vorteil, verringerte die Koloniebildung und reduzierte die Invasion — er setzte effektiv die von PPP1R14C gelöste Bremse wieder. Zusammengenommen skizziert die Studie eine klare Ereigniskette: KLF7 schaltet PPP1R14C ein; PPP1R14C neutralisiert PP1; CDK1 bleibt hyperaktiv; und Plattenepithelzellen der Lunge teilen sich unkontrolliert. Für Laien bedeutet das: Die Forschenden haben sowohl eine Warnmarke identifiziert — hohes PPP1R14C, das gefährlichere Tumoren kennzeichnet — als auch einen vielversprechenden Ansatz für Therapien: Wirkstoffe, die CDK1 hemmen, insbesondere bei Patientinnen und Patienten, deren Tumore von diesem entgleisten Kreis abhängig sind.
Zitation: Xing, L., Yuan, C., Shen, X. et al. Transcriptional activation of PPP1R14C by KLF7 unleashes CDK1 activity to promote lung squamous cell carcinoma. Sci Rep 16, 9244 (2026). https://doi.org/10.1038/s41598-026-39174-3
Schlüsselwörter: Plattenepithelkarzinom der Lunge, Zellzyklus, CDK1, PPP1R14C, zielgerichtete Therapie