Clear Sky Science · de
CiCLoDS: Gemeinsame Zellclusterbildung und Gen‑Auswahl für Einzelzell‑räumliche Transkriptomik
Nachbarschaften in der Stadt der Zellen finden
Moderne Mikroskope können inzwischen ablesen, welche Gene in Hunderttausenden von Zellen aktiv sind und dabei jede Zelle an ihrem ursprünglichen Ort im Gewebe belassen. Diese „räumliche Transkriptomik“-Revolution ist wie die Umwandlung einer verschwommenen Stadtkarte in eine Straßenniveau‑Ansicht jedes einzelnen Hauses. Allerdings gibt es einen Haken: Diese Karten enthalten Messwerte für Tausende von Genen pro Zelle — weit mehr, als Forscher leicht interpretieren oder in Folgeexperimenten messen könnten. Diese Studie stellt CiCLoDS vor, eine neue Methode, die zugleich aussagekräftige zelluläre Nachbarschaften findet und eine kleine, interpretierbare Genliste auswählt, die diese Nachbarschaften definiert.
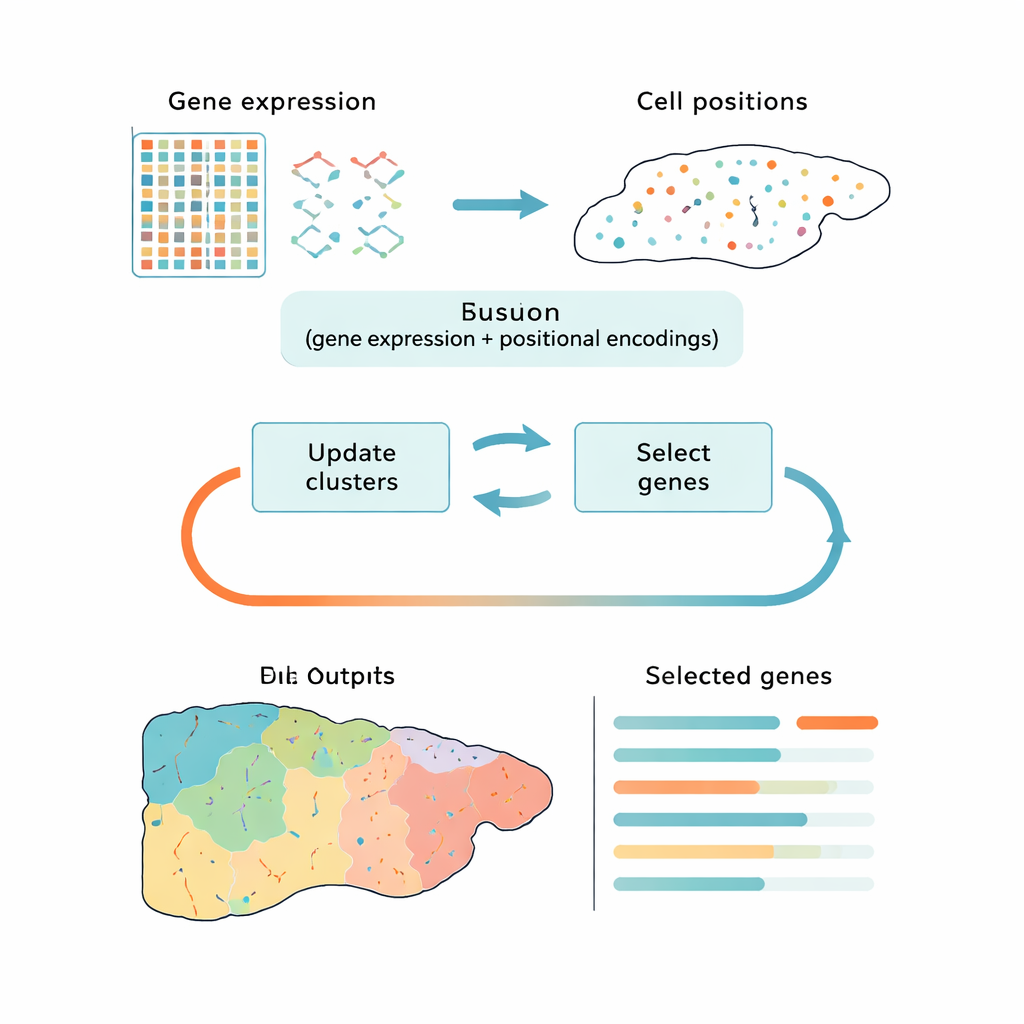
Ein klügerer Weg, große Daten zu reduzieren
Die meisten aktuellen Werkzeuge gehen dieses Problem in zwei getrennten Schritten an: Zuerst reduzieren sie die Daten auf eine einfachere Form, dann gruppieren sie Zellen in Cluster. Beliebte Ansätze wie die Hauptkomponentenanalyse (PCA) bewahren die Gesamtvarianz, können sich aber auf technische Störsignale oder generische Zellzyklus‑Signale konzentrieren statt auf die biologisch relevanten Unterschiede. Andere Methoden nutzen Deep Learning, um Muster zu finden, agieren dabei jedoch als Blackbox und geben nicht klar Auskunft darüber, welche Gene am wichtigsten sind. CiCLoDS wählt einen anderen Weg. Es behandelt Gen‑Auswahl und Clusterbildung als ein gemeinsames Problem unter einer vom Anwender definierten „Budget“-Vorgabe für die Anzahl der zu behaltenden Gene. Effektiv fragt die Methode: Welche begrenzte Menge an Genen erklärt am besten, wie Zellen in unterschiedliche Gruppen fallen, unter Berücksichtigung ihrer Genaktivität und — falls verfügbar — ihrer physischen Positionen im Gewebe?
Von der Mathematik zu Karten realer Gewebe
Die Autoren passen eine Familie mathematisch transparenter Techniken, die sogenannte Subspace‑Clustering, an die Realitäten der räumlichen Transkriptomik an, in der Datensätze über eine Million Zellen enthalten können. CiCLoDS arbeitet auf einer einfachen Zell‑nach‑Gen‑Tabelle, weist Zellen Clustern zu und bewertet gleichzeitig jedes Gen danach, wie sehr es zur Trennung dieser Cluster beiträgt. Es kann auch räumliche Informationen einweben, indem Positions‑"Encodings" hinzugefügt werden, die beschreiben, wo sich jede Zelle im Gewebe befindet, ohne die Kernoptimierung zu verändern. Auf großen Mausleber‑ und menschlichen Kolon‑Datensätzen, erzeugt von hochauflösenden Bildgebungsplattformen, läuft CiCLoDS innerhalb von Minuten auf Standardrechnern und erzeugt kompakte Genpanels — in der Größenordnung von einigen Dutzend bis wenigen Hundert Genen — die dennoch die reichhaltige Struktur der Originaldaten einfangen.
Verborgene Zonen und Blutgefäße aufdecken
Bei der Anwendung von CiCLoDS auf Mausleber fragten die Forscher, ob die Methode bekannte „Zonations“-Muster wiederfinden kann — allmähliche Verschiebungen der Hepatozytenfunktion von einer Seite des Läppchens zur anderen. Im Vergleich zu PCA und einem führenden Gen‑Auswahlwerkzeug namens geneBasis erzeugte CiCLoDS sauberere räumliche Zonen mit schärferen Grenzen und deutlich weniger fehlzugeordneten Bereichen, wie quantitative Metriken zur Übereinstimmung mit einer Referenzkarte zeigten. Bemerkenswerterweise entdeckte CiCLoDS, wenn es mehr Gene verwenden durfte, peri‑portal‑ähnliche und peri‑zentral‑ähnliche Hepatozytengruppen, die gut mit expertendefinierten Referenzclustern übereinstimmten, obwohl es nicht über das Schlüsselmarker‑Gen AXIN2 informiert war und keine expliziten räumlichen Koordinaten erhielt. Mit hinzugefügten räumlichen Encodings lernte CiCLoDS zudem Genpanels, die für Zelloberflächen‑ und gefäßbezogene Funktionen angereichert waren, und konnte echte Blutgefäße von Bildgebungsartefakten genau unterscheiden — etwas, das einfachere Methoden entweder nicht schafften oder nur mit zusätzlichen ad‑hoc‑Anpassungen erreichten.
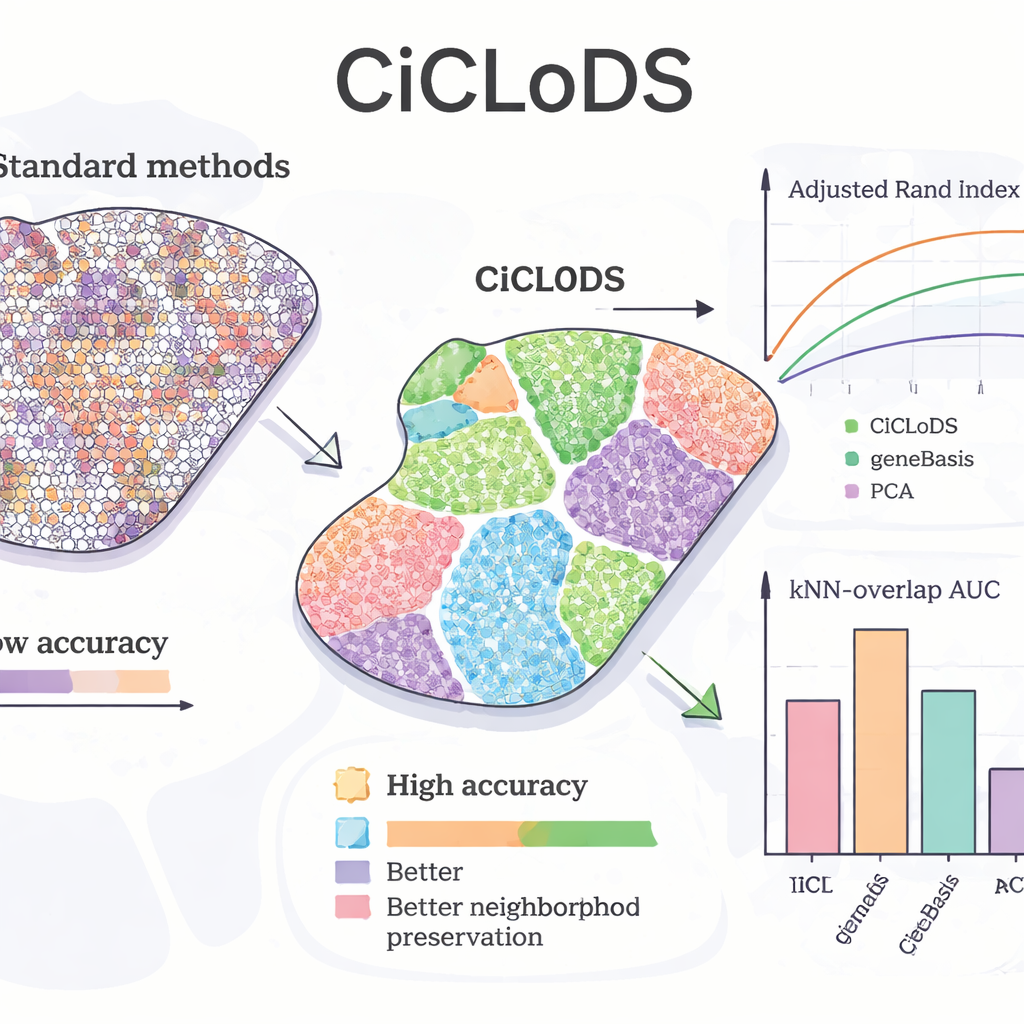
Generalisierung über Gehirne hinweg und Verstärkung anderer Methoden
Um zu prüfen, ob CiCLoDS über sehr unterschiedliche Gewebe und Individuen hinweg robust ist, analysierten die Autoren Proben des dorsolateralen präfrontalen Kortex von drei menschlichen Spendern. Hier lieferte CiCLoDS vergleichbare oder bessere Ergebnisse als spezialisierte räumliche Methoden wie BayesCafe und BayesSpace, insbesondere bei einer schwierigen Probe, bei der die anderen Werkzeuge Probleme hatten. Die Studie hebt auch eine „Hybrid“-Nutzung hervor: Zuerst CiCLoDS laufen lassen, um stabile Cluster zu erhalten, und diese dann in BayesSpace einspeisen. Diese Warm‑Start‑Strategie erhöhte die Gesamtgenauigkeit und erzeugte Schichtmuster des Gehirns, die am besten mit Expertenannotationen übereinstimmten, und zeigt, dass CiCLoDS sowohl eigenständig funktionieren als auch nachgelagerte probabilistische Modelle zuverlässiger machen kann.
Warum das für Biologie und Medizin wichtig ist
Für Nicht‑Spezialisten ist die Kernbotschaft: CiCLoDS verwandelt überwältigende Zellkarten in prägnante, biologisch sinnvolle Zusammenfassungen. Statt mit Tausenden rauschbehafteter Messwerte zu arbeiten, erhalten Forschende eine handhabbare Genliste und klare räumliche Cluster, die reale Gewebeorganisation widerspiegeln — etwa Stoffwechselzonen in der Leber, Blutgefäße und ihre Nischen sowie geschichtete Strukturen im Gehirn. Da das Genbudget benutzerkontrolliert ist und die Berechnungen ressourcenschonend sind, kann CiCLoDS helfen, gezielte Genpanels für zukünftige Experimente zu entwerfen, die Interpretation komplexer räumlicher Datensätze zu leiten und robuste Ausgangspunkte für aufwändigere Modellierungen zu liefern. In einer Ära, in der die Flaschenhals nicht mehr die Datenerhebung, sondern das Verständnis ist, versprechen Werkzeuge wie CiCLoDS, hochdimensionale Gewebekarten sowohl praktikabel als auch erkenntnisreich zu machen.
Zitation: Wang, N., He, Y., Ray, E. et al. CiCLoDS: Joint cell clustering and gene selection for single-cell spatial transcriptomics. Sci Rep 16, 5356 (2026). https://doi.org/10.1038/s41598-026-39168-1
Schlüsselwörter: räumliche Transkriptomik, Zellclusterbildung, Auswahl von Genpanels, Gewebearchitektur, Einzelzellanalyse