Clear Sky Science · de
Bewertung der evolutionären Beziehung des TATA-Bindungsproteins (TBP) zu verschiedenen Faltungsmustern von Proteindomänen mithilfe von Support Vector Machine (SVM)
Wie ein DNA‑„An‑Schalter“-Protein mit vielen anderen verbunden ist
Das TATA‑Box‑Bindeprotein, kurz TBP, ist ein Arbeitstier unserer Zellen: Es hilft, Gene einzuschalten, indem es an viele Promotoren die DNA ergreift. Diese Studie stellt eine täuschend einfache Frage mit weitreichenden Folgen: Gibt es andere Proteine mit sehr unterschiedlichen Aufgaben, die heimlich TBPs zugrunde liegende Form teilen? Durch die Kombination von 3D‑Strukturvergleichen, Sequenzanalyse und modernen maschinellen Lernverfahren verfolgen die Autoren verborgene Verwandtschaften zwischen TBP und Proteinen, die an Stoffwechselprozessen, Neurotransmitterchemie und sogar krebsrelevanten Signalwegen beteiligt sind.
Ein zentrales Protein im Zentrum der Genkontrolle
TBP sitzt am Eingang der Genexpression in Organismen von Hefen bis zum Menschen. Es erkennt eine kurze DNA‑Sequenz, die TATA‑Box genannt wird, und krümmt die DNA, um den Aufbau der großen Transkriptionsmaschinerie zu erleichtern, die Gene in RNA umschreibt. Da dieser Schritt so zentral ist, ist die Faltung — die dreidimensionale Anordnung — des TBP‑Kerns über die Evolution hinweg hoch konserviert. Die Autoren konzentrieren sich auf eine gut untersuchte TBP‑Struktur, bekannt als 1tba, und verwenden sie als Sonde, um nach anderen Proteinen zu suchen, die möglicherweise denselben architektonischen Bauplan teilen, selbst wenn ihre Aminosäuresequenzen und Alltagsfunktionen auf den ersten Blick sehr unterschiedlich erscheinen.
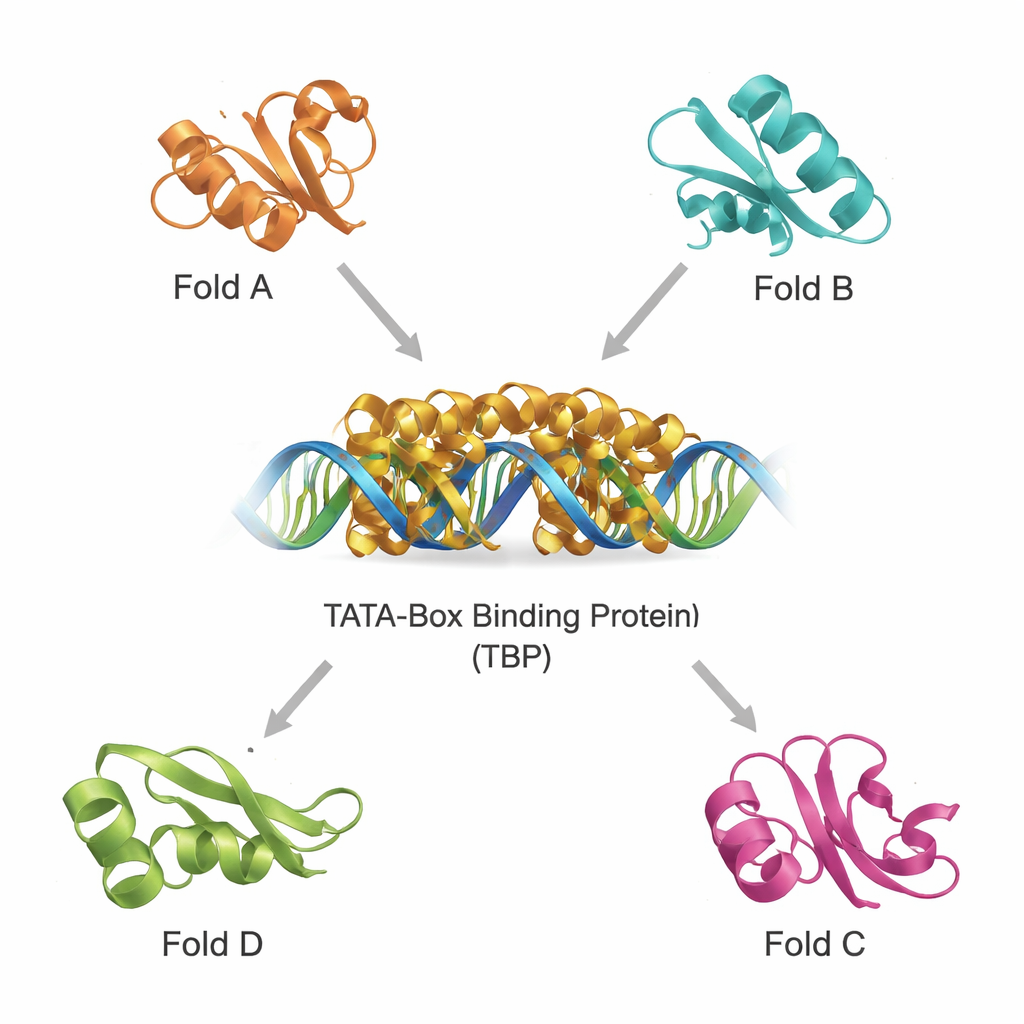
Strukturelle Verwandte in einem dicht besetzten Proteinuniversum finden
Moderne Datenbanken enthalten Hunderttausende von Proteinstrukturen, sodass sich entfernte Verwandte eher nach 3D‑Form als allein nach Sequenz suchen lassen. Mithilfe zweier leistungsfähiger Werkzeuge, DALI und TOP‑search, filterte das Team zunächst Proteine heraus, deren Faltungen TBP ähnlich sahen. Anschließend klassifizierten sie diese Kandidaten mithilfe eines evolutionären Domänenkatalogs und reduzierten die Auswahl auf eine kleine Gruppe strukturell ähnlicher, aber funktional vielfältiger Beispiele. Dazu gehören ein für den Stoffwechsel wichtiger Glutamin‑bildender Enzymtyp, eine Domäne, die in mehreren tRNA‑bearbeitenden Enzymen vorkommt, ein Enzym mit charakteristischer „Hot‑Dog“-Faltung, das an Fettsäurechemie beteiligt ist, und Proteine, die an der Herstellung von Tetrahydrobiopterin mitwirken, einem für die Gehirnfunktion wesentlichen Molekül. Überlagerungen ihrer Strukturen mit TBP zeigten, dass sie trotz unterschiedlicher Aufgaben erkennbare Kernmotive teilen.
Maschinen beibringen, versteckte Proteinfamilien zu erkennen
Um über Einzelfallbetrachtungen hinauszugehen, bauten die Autoren maschinelle Lernmodelle, die automatisch TBP‑ähnliche Faltungen markieren können. Sie stellten große Datensätze von Proteinsequenzen zusammen, die entweder zu TBP oder zu jedem der verwandten Faltungstypen gehören, sowie eine breite „Hintergrund“-Menge unzusammenhängender Proteine. Jedes Protein wurde in einfache numerische Zusammenfassungen überführt: wie häufig jede Aminosäure vorkommt und wie häufig jedes mögliche Paar von Aminosäuren in der Sequenz auftritt. Diese Profile wurden in Support Vector Machines (SVMs) und Random‑Forest‑Modelle eingespeist, die lernten, einen Faltungstyp von allen anderen zu unterscheiden. Mit rigoroser Kreuzvalidierung erreichten die Modelle sehr hohe Genauigkeiten — oft über 95 Prozent — selbst wenn sie nur mit Sequenzabschnitten trainiert wurden, die konservierten Regionen entsprechen.
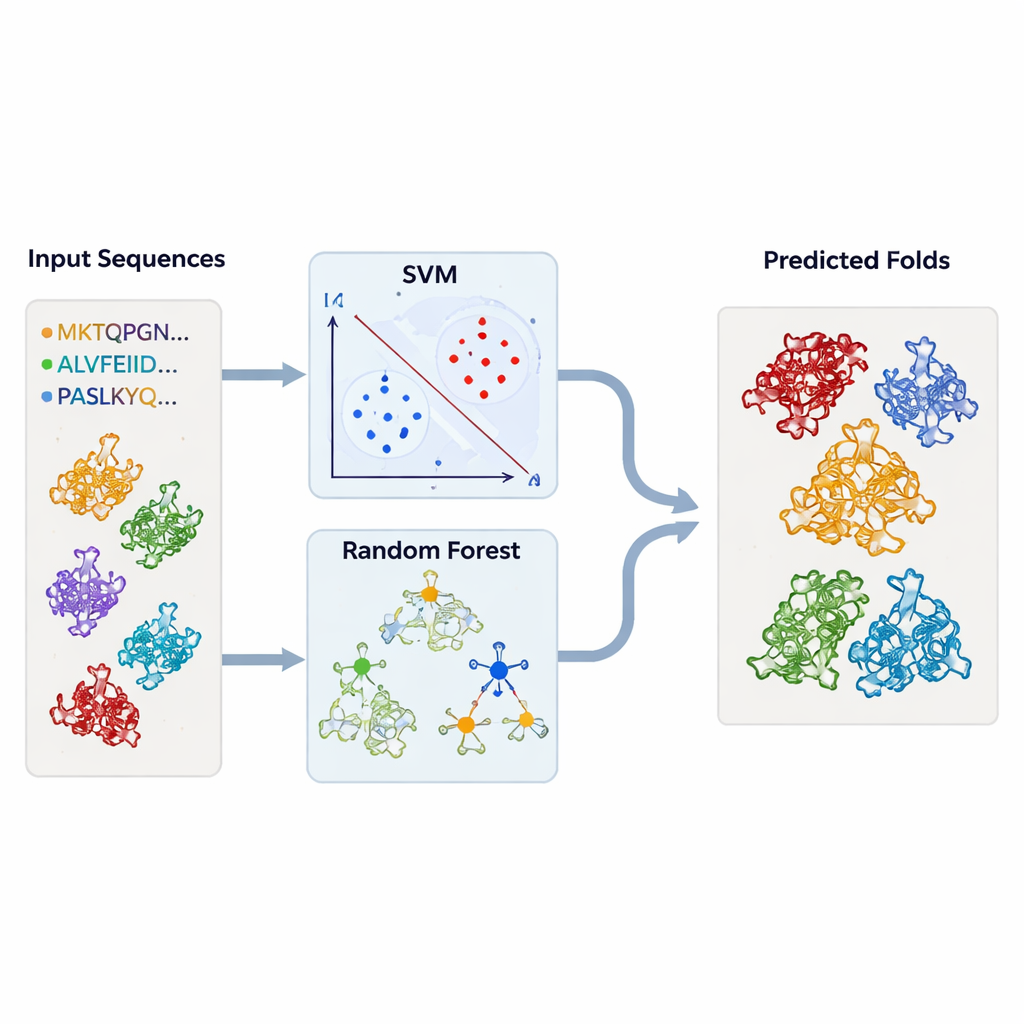
Die Modelle an Tausenden unbekannter Strukturen testen
Mit diesen trainierten Klassifikatoren kehrte das Team zu den Strukturdatensätzen zurück. Sie führten Tausende von Proteinketten — aus DALI und TOP‑search gewonnen — durch ihre Modelle, um zu sehen, welche die statistischen Kennzeichen TBP‑ähnlicher oder verwandter Faltungen trugen. Die SVM‑ und Random‑Forest‑Ansätze stimmten größtenteils überein und identifizierten viele Kandidaten, die auch von strukturellen Werkzeugen als ähnlich markiert worden waren. In einigen Fällen gruppierten sich Enzyme mit scheinbar unzusammenhängenden Aktivitäten dennoch stark mit TBP oder untereinander, was die Idee untermauert, dass die Evolution denselben zugrunde liegenden Rahmen für viele unterschiedliche biochemische Aufgaben wiederverwenden kann.
Warum diese verborgenen Verbindungen wichtig sind
Die Studie kommt zu dem Schluss, dass TBP eine tiefe strukturelle Abstammung mit mehreren Enzymfamilien teilt, darunter Glutamin‑Synthetase‑ähnliche Proteine und Editierdomänen von tRNA‑verarbeitenden Enzymen. Selbst wenn Sequenzen auseinandergedriftet und Funktionen divergiert sind, behalten diese Proteine gemeinsame Architekturmotive bei, was auf Abstammung von einem gemeinsamen Vorfahren hindeutet. Für einen Nicht‑Spezialisten lautet die Kernbotschaft: Die Natur neigt dazu, erfolgreiche Entwürfe wiederzuverwenden — eine Faltung kann wiederholt angepasst werden, um sehr unterschiedliche Probleme zu lösen, vom Einschalten von Genen bis zur Feinabstimmung von Stoffwechsel und Gehirnchemie. Durch die Kombination von 3D‑Strukturvergleich und maschinellem Lernen liefern die Autoren ein praktisches Werkzeugset, um solche Beziehungen aufzudecken, Biologen bei der Vorhersage der Funktion uncharakterisierter Proteine zu unterstützen und Pharmaforscher auf neue, evolutionär geleitete Zielstrukturen in krankheitsrelevanten Wegen hinzuweisen.
Zitation: Selvaraj, M.K., Kaur, J. Evaluating the evolutionary relationship of TATA binding protein (TBP) with various folding patterns of protein domains using support vector machine (SVM). Sci Rep 16, 7696 (2026). https://doi.org/10.1038/s41598-026-38883-z
Schlüsselwörter: TATA-Box-Bindeprotein, Protein-Evolution, Maschinelles Lernen, Proteinstruktur, Support Vector Machine