Clear Sky Science · de
Computergestützte Optimierung der Löslichkeit der Calpain-Domäne von DEK1 durch integrierte Strukturmodellierung und datengetriebene gezielte Mutagenese
Warum es wichtig ist, pflanzliche Proteine gefügig zu machen
Viele der Proteine, die das Pflanzenwachstum steuern, sind große, empfindliche Moleküle, die sich im Labor nicht auflösen lassen, wenn Forschende sie untersuchen wollen. Ein solches Protein, DEK1, hilft, die Gestalt von Pflanzen von der Einzelzelle bis zur Gesamtstruktur zu formen. Da jedoch ein entscheidender Teil von DEK1 bei der Produktion in Bakterien verklumpt, blieb seine 3D-Struktur unbekannt, was die Bemühungen, es zu verstehen und zu nutzen, verlangsamte. Diese Studie zeigt, wie Computer-Modellierung und intelligente, datengetriebene Gestaltung jenes problematische Bereichs so verändern können, dass er besser löslich wird, ohne seine grundlegende Architektur zu zerstören — und liefert damit eine allgemeine Anleitung zum Zähmen schwieriger Proteine.
Den Problembereich eines Schlüsselproteins ins Visier nehmen
DEK1 ist ein ungewöhnlich großes Protein, das in Zellmembranen verankert ist und an einem Ende eine schneidende Enzymregion trägt, die als Calpain-Domäne bezeichnet wird. Genetische Studien haben gezeigt, dass diese Domäne für eine normale Entwicklung von Pflanzen wie Moosen und Kulturpflanzen unerlässlich ist, doch ihre Struktur wurde nie experimentell aufgeklärt. Wenn Forschende versuchen, diesen Calpain-Kern (CysPc genannt) im üblichen Wirtsbakterium Escherichia coli herzustellen, neigt er dazu, unlöslich zu werden und dichte Einschlusskörper zu bilden. Das macht es nahezu unmöglich, ihn in den Mengen und der Qualität zu reinigen, die für detaillierte Struktur- und Funktionsstudien nötig sind. Die Autorinnen und Autoren setzten sich daher zum Ziel, die CysPc-Domäne so umzudesignen, dass sie sich leichter auflöst, ohne ihre Gesamtform zu verlieren.
Ein vertrauenswürdiges 3D-Modell von Grund auf erstellen
Weil keine experimentelle Struktur für dieses pflanzliche Calpain vorlag, musste das Team zunächst seine 3D-Form vorhersagen. Sie kombinierten mehrere moderne Strukturvorhersagewerkzeuge, darunter AlphaFold2, SWISS-MODEL und I-TASSER, und verankerten diese Vorhersagen an bekannten Strukturen verwandter tierischer Calpaine. Mit einem Konsensusansatz verfeinerten und überprüften sie die resultierenden Modelle mit mehreren Qualitätstests, die Rückgratgeometrie, Packung und Übereinstimmung mit bekannten Strukturmustern bewerten. Diese unabhängigen Kontrollen zeigten, dass das integrierte Modell der CysPc-Domäne verlässlicher war als jede einzelne Vorhersage allein und eine solide Ausgangsbasis bot, um zu untersuchen, wie kleine Änderungen der Aminosäuresequenz die Löslichkeit verbessern könnten.
Virtuelle Mutationen im simulierten Lösungsmittel testen
Mit dem 3D-Modell in der Hand führten die Autorinnen und Autoren umfangreiche Molekulardynamik-Simulationen durch, bei denen das Protein und die umgebenden Wassermoleküle im Computer über die Zeit verfolgt werden. Sie konzentrierten sich auf Residuen an der Proteinoberfläche, die flexibel, hydrophob oder als förderlich für Aggregation vorhergesagt waren. Kandidatenpositionen wurden einzeln in wasserfreundlichere Aminosäuren verwandelt und jeweils für 200 Nanosekunden simuliert. Für jede Variante maßen sie Merkmale, die mit Löslichkeit zusammenhängen, etwa wieviel Oberfläche mit Wasser in Kontakt steht, wie kompakt das Protein bleibt und wie stark Atome schwanken. Viele Einzelmutationen erhöhten moderat die Lösungsmittelexposition oder interne Wasserstoffbrücken, während die Gesamtfaltung erhalten blieb, was darauf hindeutet, dass das Grundgerüst von CysPc sorgfältig ausgewählte Substitutionen tolerieren könnte.
Algorithmen die Mutationsräume durchsuchen lassen
Die Veränderung nur einer Aminosäure führt selten zu großen Verbesserungen der Löslichkeit, weshalb die Forschenden als Nächstes Kombinationen aus zwei und drei Mutationen untersuchten. Sie erzeugten eine Bibliothek von Doppel- und Dreifachvarianten, aufgebaut aus den besten Einzelmutationen, und simulierten jede davon erneut. Um diese Designs fair zu bewerten und zu reihen, definierten sie einen gewichteten Index, der mehrere Simulationsmerkmale kombiniert, die bekanntermaßen mit Löslichkeit korrelieren, und belohnte erhöhte Hydratation und interne Bindungen, während übermäßige Flexibilität bestraft wurde. Anschließend nutzten sie einen Reinforcement-Learning-Algorithmus (Proximal Policy Optimization), um den riesigen Raum möglicher Dreifachmutanten zu durchforsten und die vielversprechendsten Kombinationen vorzuschlagen. Diese datengetriebene Suche konvergierte auf einen bestimmten Dreifachmutanten, genannt MUT347, als Top-Kandidat.
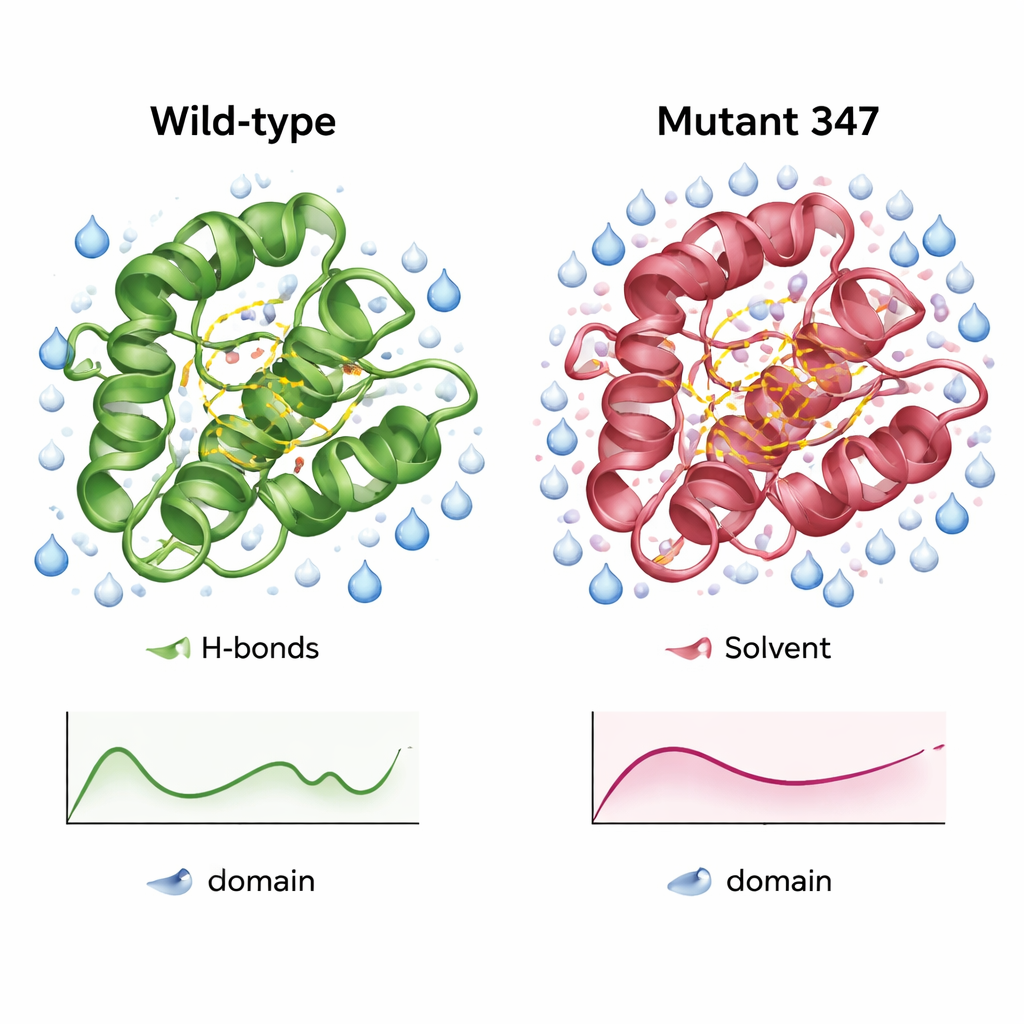
Eine kompaktere, besser hydratisierte Version des Enzyms
Detaillierte Simulationen des Wildtyp-CysPc und von MUT347 zeigten, worin sich die designte Variante unterschied. MUT347 equilibrierte schneller und wies insgesamt kleinere Abweichungen von seiner Ausgangsform auf, was auf eine höhere strukturelle Stabilität in Lösung hindeutet. Seine Schleifen und Kettenenden waren etwas weniger schlaff, während der katalytische Kern seine ursprüngliche Beweglichkeit behielt, was darauf schließen lässt, dass funktionell wichtige Bewegungen erhalten blieben. Der Dreifachmutant zeigte mehr interne Wasserstoffbrücken und an Schlüsselstellen eine größere wasserzugängliche Oberfläche — Zeichen einer besser organisierten und stärker hydratisierten Oberfläche. Unter variierenden Salzkonzentrationen und pH-Werten hielt MUT347 durchweg geringere Fluktuationen als das ursprüngliche Protein, ein Verhalten, das mit einer verringerten Neigung zur Aggregation einhergeht.
Was das für das Studium und die Wiederverwendung von Proteinen bedeutet
Für Nicht-Spezialisten lautet die Schlussfolgerung: Die Autorinnen und Autoren haben ein weitgehend computergestütztes Rezept entwickelt, um ein schwieriges, verklumpendes Fragment eines wichtigen Pflanzenproteins in eine besser lösliche, gutmütigere Version zu verwandeln, ohne die Struktur zuvor experimentell bestimmen zu müssen. Durch die Kombination moderner Strukturvorhersage, langzeitiger Simulationen und lernender Algorithmen, die viele Designentscheidungen gleichzeitig abwägen können, identifizierten sie eine Dreifachmutation, die die Faltung stabilisieren und die Oberfläche günstiger dem Wasser aussetzen soll. Zwar sind experimentelle Untersuchungen weiterhin nötig, um die Verbesserungen in echten Reagenzgläsern zu bestätigen, doch könnte dieses Rahmenkonzept breit anwendbar sein, um andere eukaryotische Proteine zu retten, die schwer herzustellen sind, und Forschenden so helfen, Strukturen und Funktionen freizulegen, die derzeit unerreichbar sind.
Zitation: Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep 16, 7767 (2026). https://doi.org/10.1038/s41598-026-38805-z
Schlüsselwörter: Proteinlöslichkeit, computergestützte Mutagenese, molekulardynamik, pflanzliches Calpain DEK1, Proteinengineering