Clear Sky Science · de
LncRNA FTX fördert Myokardfibrose, indem sie miR-335-3p bindet und dadurch die TFEC/ILK-Signalgebung reguliert
Warum Vernarbung des Herzens wichtig ist
Herzinsuffizienz betrifft weltweit zig Millionen Menschen und entwickelt sich oft über Jahre hinweg still und schleichend. Ein zentraler Treiber dieses Funktionsverlusts ist die Myokardfibrose — die langsame, progressive Vernarbung des Herzmuskels, die ihn steifer macht und seine Pumpleistung verringert. Diese Studie untersucht die molekulare "Verdrahtung", die Zellen im Herzen dazu bringt, zu viel Narbengewebe zu bilden, und identifiziert eine neue Signalkette von Molekülen, die gezielt werden könnte, um diesen schädlichen Prozess zu verlangsamen oder sogar umzukehren.
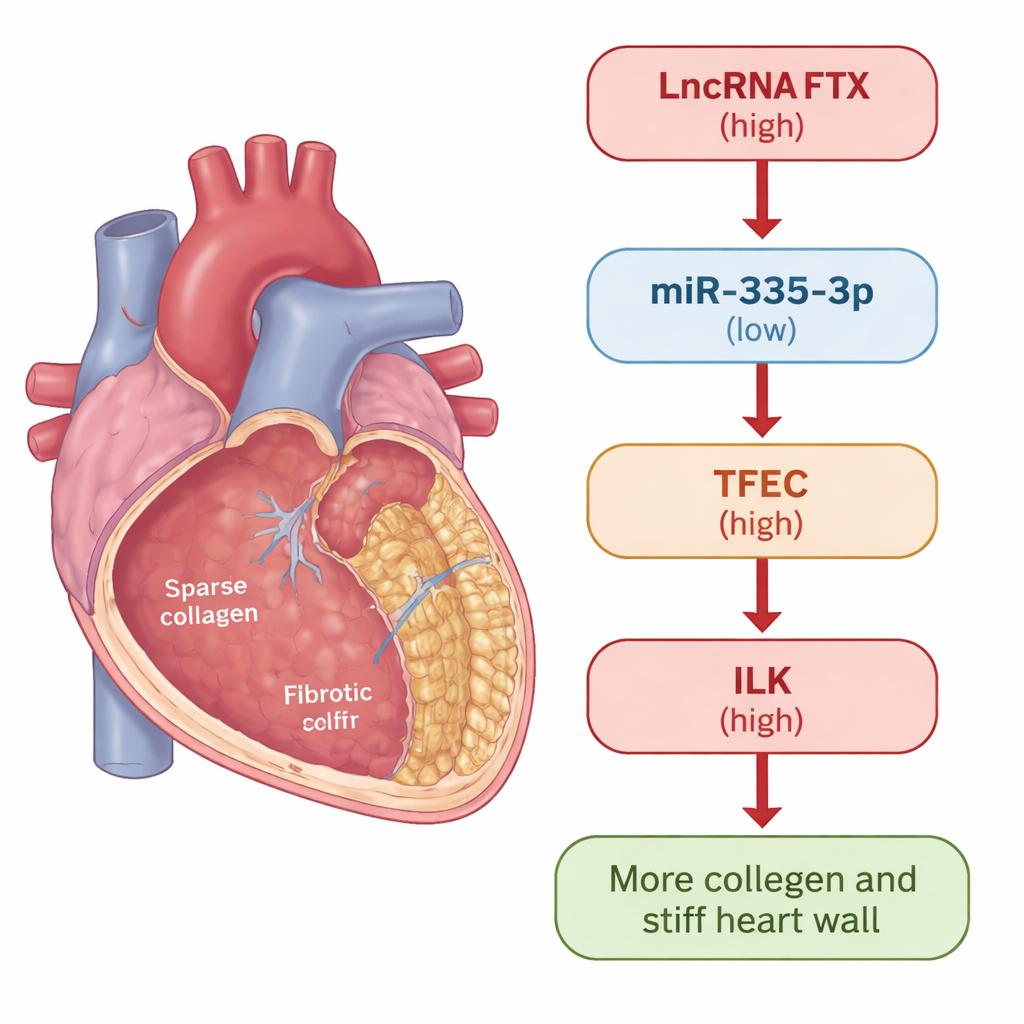
Ein genauerer Blick auf Herzvernarbung
Wenn das Herz verletzt wird oder unter Stress steht, treten sogenannte kardiale Fibroblasten in Aktion. Bei einer gesunden Reparatur helfen sie, Schäden zu verschließen. In chronischen Erkrankungen können sie jedoch in einen überaktiven Zustand umschalten und überschüssiges Kollagen sowie andere Bestandteile der extrazellulären Matrix produzieren, wodurch die Herzwand schließlich versteift. Die Forschenden verwendeten zwei Modelle, um diesen Prozess zu untersuchen: Mäuse, die mit dem Wirkstoff Isoproterenol behandelt wurden, wodurch zuverlässig Herzfibrose ausgelöst wird, und humanes kardiales Fibroblastenmaterial, das dem Signalmolekül TGF-β1 ausgesetzt wurde, einem bekannten Auslöser für Vernarbung. In beiden Systemen maßen sie, wie sich bestimmte Gene und Proteine während der Entwicklung der Fibrose veränderten.
Eine schädliche Kettenreaktion in Zellen
Das Team konzentrierte sich auf einen Transkriptionsfaktor namens TFEC, ein Protein, das im Zellkern sitzt und andere Gene anschaltet. Sie stellten fest, dass TFEC zusammen mit einem weiteren Protein, der integrin-gekoppelten Kinase (ILK), konsistent erhöht war, wenn Fibroblasten in einen fibrotischen, narbenbildenden Zustand gedrängt wurden. Das Stilllegen von TFEC oder ILK verringerte deutlich klassische Fibrosemarker wie α-glattes Muskelactin und die Kollagene I und III und dämpfte außerdem einen wachstumsregulierenden Signalweg (Akt/GSK3β und Hippo-Signalgebung), der für Gewebsvernarbung bekannt ist. DNA-Bindungsexperimente zeigten, dass TFEC direkt an den Promotor des ILK-Gens bindet und dessen Aktivität steigert, wodurch TFEC eindeutig oberhalb von ILK in einer pro-fibrotischen Signalkaskade eingeordnet wird.
RNA-Schalter, die den Master-Regulator steuern
Um zu verstehen, was TFEC selbst kontrolliert, betrachteten die Forschenden nichtkodierende RNAs — RNA-Moleküle, die keine Proteine erzeugen, aber als Feinregulatoren der Genaktivität fungieren. Sie identifizierten eine kleine RNA, miR‑335‑3p, die in fibrotischen Herzen und Zellen reduziert war. Eine Erhöhung von miR‑335‑3p senkte TFEC, während dessen Blockade TFEC erhöhte; Reportertests bestätigten, dass miR‑335‑3p direkt an TFEC-Botschaften bindet und diese in Schach hält. Anschließend fanden sie eine lange nichtkodierende RNA namens FTX, die in der Fibrose erhöht war und physisch mit miR‑335‑3p interagierte. FTX verhielt sich wie ein molekularer Schwamm: Es band miR‑335‑3p und hinderte diese kleine RNA daran, TFEC zu bremsen. Infolgedessen stiegen TFEC und ILK an, und die Fibroblasten produzierten mehr narbenbildendes Kollagen.
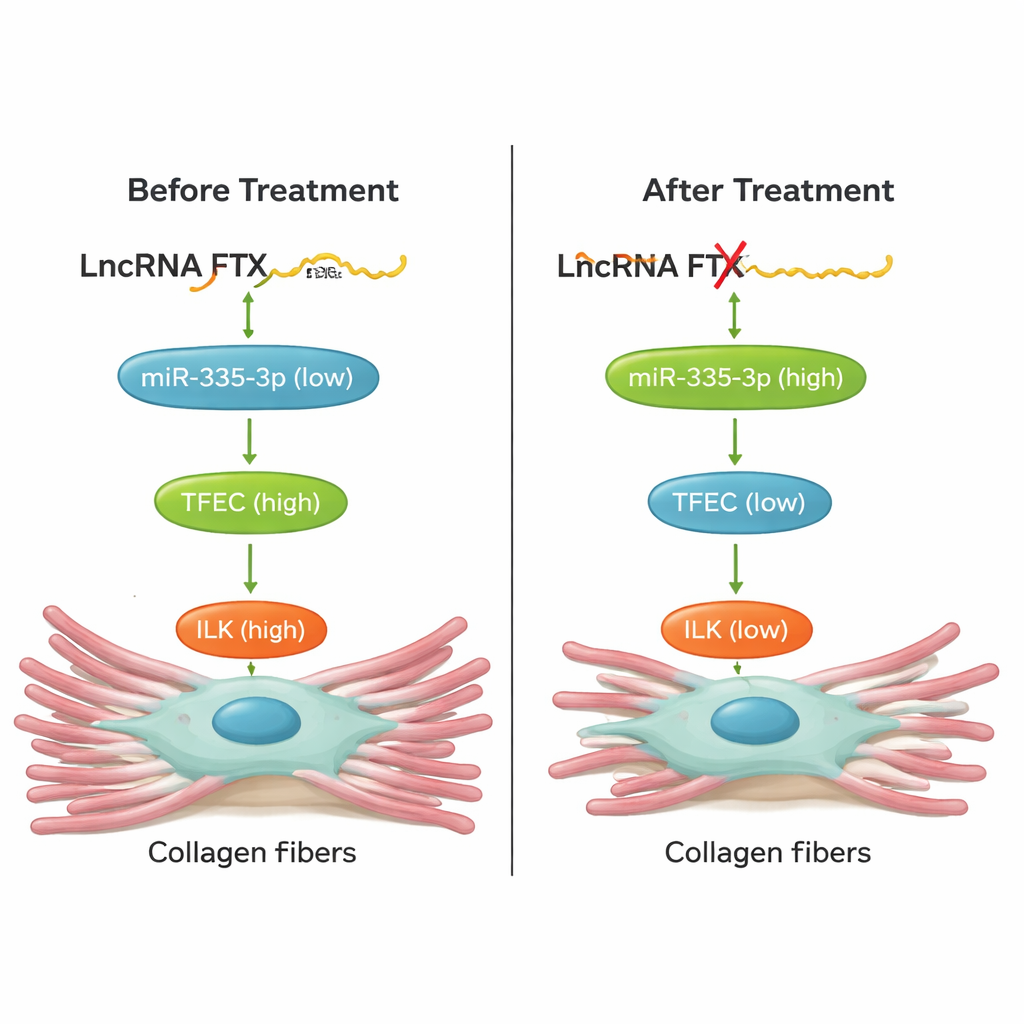
Vom Zellkulturmodell zum lebenden Herzen
Wesentlich ist, dass das Team prüfte, ob die Störung dieser Kette tatsächlich Herzen in Tieren schützen kann. Bei Mäusen, die Isoproterenol ausgesetzt waren, führten das Herunterregulieren von TFEC, das Herabsetzen von FTX im Herzen mittels eines AAV9-Gentherapie-Vektors oder das Erhöhen von miR‑335‑3p mit einem chemisch stabilisierten "Agomir" alle zu weniger Kollagenablagerung und niedrigeren Spiegeln von Fibrosemarkern im Herzgewebe. Diese Interventionen verbesserten außerdem die Herzfunktion: Schlagvolumen und Ejektionsfraktion bewegten sich wieder in Richtung normal, und schädliche Erhöhungen der Herzfrequenz wurden abgefedert. Rescue-Experimente in Zellen zeigten, dass die Veränderung einer Komponente der FTX/miR‑335‑3p/TFEC/ILK-Achse die anderen vorhersehbar verschob, was bestätigt, dass es sich um einen eng verknüpften Signalweg und nicht um eine lose Korrelation handelt.
Was das für künftige Therapien bedeutet
Für Nichtfachleute lautet die Kernaussage: Die Autorinnen und Autoren haben einen neuen "Kontrollhebel" für Herzvernarbung identifiziert. Eine lange RNA namens FTX hebt die Bremse (miR‑335‑3p) von einem Master-Schalter (TFEC), der daraufhin ILK und nachgelagerte pro-fibrotische Signale aktiviert, was zu überschüssiger Kollagenablagerung und Versteifung des Herzens führt. Durch das Reduzieren von FTX, das Wiederherstellen von miR‑335‑3p oder das direkte Blockieren von TFEC war es in Mäusen möglich, Vernarbung zu verringern und die Pumpfunktion zu verbessern. Zwar sind weitere Untersuchungen erforderlich, um diesen Weg beim Menschen zu bestätigen und sichere Therapien zu entwickeln, doch bietet diese RNA-basierte Regulationskette mehrere vielversprechende Angriffspunkte, um fibrosegetriebener Herzinsuffizienz entgegenzuwirken.
Zitation: Yao, F., He, Z., Zheng, C. et al. LncRNA FTX promotes myocardial fibrosis by sponging miR-335-3p to regulate TFEC/ILK signaling. Sci Rep 16, 7340 (2026). https://doi.org/10.1038/s41598-026-38615-3
Schlüsselwörter: Myokardfibrose, Herzinsuffizienz, nichtkodierende RNA, kardiale Fibroblasten, Fibrose-Signalgebung