Clear Sky Science · de
Optimierung von Galectin‑3‑Bindungspartnern durch in situ-Synthese mehrerer Verbindungen und native Massenspektrometrie
Warum das für zukünftige Medikamente wichtig ist
Viele moderne Arzneimittel wirken, indem sie an Proteine im Körper binden. Ein kleines Molekül zu finden, das genau und stark an die richtige Stelle bindet, ist jedoch langsam, teuer und oft frustrierend. Diese Studie stellt eine schnellere Methode vor, um solche Moleküle direkt in Anwesenheit des Zielproteins zu verfeinern und die besten Kandidaten anschließend mit einer hochempfindlichen Wägungstechnik zu identifizieren. Die Autoren demonstrieren ihren Ansatz an Galectin‑3, einem Protein, das mit Krebswachstum in Verbindung steht, und erhalten letztlich einen vielversprechenden, arzneimittelähnlichen Kandidaten, der so stark bindet wie einige der besten bestehenden Verbindungen, jedoch an einer unerwarteten Tasche auf der Proteinoberfläche.

Neues Denken bei der Suche nach besseren Wirkstoff‑Leads
Traditionelle Wirkstoffoptimierung gleicht einem teuren Ratespiel. Chemiker verändern eine Ausgangsverbindung Schritt für Schritt, testen jede Version und hoffen, die Bindungsstärke zum Zielprotein zu verbessern. Proteinoberflächen sind jedoch flexibel, Wassermoleküle stören, und das Bindungsereignis selbst kann das Protein umgestalten, sodass Computervorhersagen unzuverlässig werden. Selbst bei einer hochauflösenden Struktur gibt es keine Garantie, dass eine vorgeschlagene Änderung nützlich ist. Bestehende "target‑guided" Methoden versuchen zwar, dem Protein seine Partner aus einem Bausteinpool wählen zu lassen, doch diese Ansätze sind weiterhin auf komplexe Analysen und indirekte Signale angewiesen, um zu schließen, welche Verbindung tatsächlich am besten bindet.
Das Protein entscheiden lassen – und dann die Gewinner wiegen
Die Forscher kombinierten zwei Ideen zu einem schlanken Workflow. Zunächst nutzten sie eine reversible chemische Reaktion, die einen gemeinsamen zuckerbasierten Kern mit vielen verschiedenen Seitengruppen in einem einzigen Gefäß verknüpft und so ein Gemisch verwandter Moleküle bildet. Durch sorgfältige Anpassung der Ausgangsverhältnisse gelangen die Produkte in ein ausgeglichenes Gleichgewicht, das durch einfache Konzentrationsregeln gesteuert wird und ihre Mengen trotz unterschiedlicher Rohreaktivitäten angleicht. Zweitens setzten sie dieses Gemisch Galectin‑3 aus und untersuchten es mittels nativer Massenspektrometrie, einer Form der Massenspektrometrie, die Protein‑Molekül‑Paare in einer schonenden, wasserähnlichen Lösung intakt hält. Da jeder Kandidat eine eindeutige Masse besitzt, kann das Instrument direkt erkennen, welche Moleküle tatsächlich an das Protein gebunden sind – ganz ohne Marker oder Referenzen.
Vom überfüllten Gemisch zu einem herausragenden Binder
Mit diesem Aufbau stellte das Team Dutzende Galectin‑3‑Binder her, indem verschiedene Seitengruppen an einen Zuckerkern angehängt wurden, der von einem bekannten Inhibitor, GB1107, inspiriert ist. Sie teilten 35 verschiedene Hydrazid‑Bausteine in handhabbare Gruppen, bildeten alle Kombinationen in situ und fügten dann Galectin‑3 hinzu. Die native Massenspektrometrie hob jene Verbindungen hervor, die am häufigsten zusammen mit dem Protein vorkamen und markierte sie als primäre Treffer. Ein nachfolgender Thermostabilitätstest – der misst, wie eine Verbindung das Protein beim Erhitzen stabilisiert – sortierte Falschpositive aus, die durch Besonderheiten der Gasphasenmessung entstanden. Drei führende Kandidaten blieben übrig, und detaillierte wärmebasierte Bindungsmessungen zeigten, dass einer von ihnen, genannt GalAldBZ20, Galectin‑3 besonders stark gebunden hat, im submikromolaren Bereich.
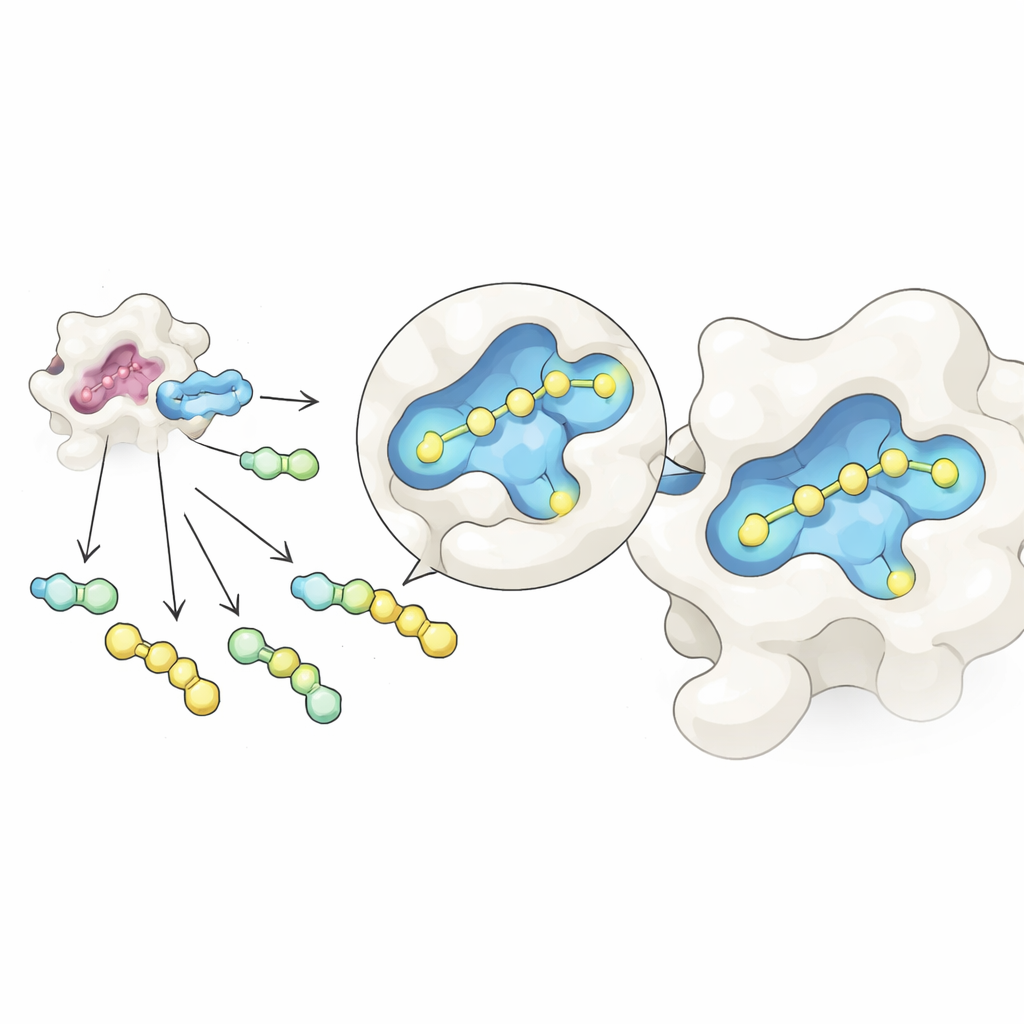
Eine verborgene Tasche entdecken und verstärken
Die nächste Überraschung ergab sich, als das Team untersuchte, wie GalAldBZ20 auf der Oberfläche von Galectin‑3 sitzt. Die meisten bekannten Binder nutzen eine "alpha"‑Tasche in der Nähe der Zuckerbindungsstelle, doch Strukturdaten und Computersimulationen deuteten darauf hin, dass GalAldBZ20 stattdessen eine benachbarte "beta"‑Tasche bevorzugt. Röntgenkristallographie lieferte Hinweise darauf, Kernspinresonanz im Lösungsmittel zeigte mehrere lokale Konformationen in der Nähe dieser Tasche, und Molekulardynamik‑Simulationen stützten ein Modell, in dem ein nitro‑haltiger Ring des Moleküls in der Beta‑Stelle Platz nimmt. In der Annahme, dass sich diese Anordnung fester verankern lässt, überarbeiteten die Chemiker den chemischen Linker zwischen Zucker und Nitro‑Ring, um neue polare Kontakte mit dem Protein zu fördern und die Flexibilität zu verringern.
Aus einem cleveren Screening einen starken Kandidaten formen
Mit dieser Erkenntnis synthetisierte das Team eine kleine Reihe rigiderer Folge‑Moleküle, die denselben Zucker und Nitro‑Ring beibehielten, aber den Verbindungsabschnitt zwischen ihnen veränderten. Eine Variante, ein N‑Galactosid (Verbindung 5), stach heraus: Sie band Galectin‑3 etwa zehnmal stärker als der ursprüngliche Treffer und erreichte eine Bindungsstärke, die mit GB1107 vergleichbar ist, blieb dabei aber bevorzugt in der Beta‑Tasche. Eine ultrahochauflösende Kristallstruktur zeigte eine klare Elektronendichte für den Nitro‑Ring in dieser Tasche, gestützt durch mehrere Wasserstoffbrücken und eine Kation‑π‑Wechselwirkung mit wichtigen Aminosäuren. Beim Entfernen oder Ersetzen der Nitrogruppe durch eine einfache Methylgruppe schwächte sich die Bindung deutlich ab, was deren Bedeutung unterstreicht. Da Galectin‑1, ein verwandtes Protein, diese Beta‑Tasche nicht besitzt, könnte die neue Verbindung letztlich eine bessere Selektivität bieten – eine begehrte Eigenschaft im Wirkstoffdesign.
Was das für die künftige Wirkstoffforschung bedeutet
Anschaulich zeigt diese Arbeit, dass man viele verwandte Moleküle zusammenmischen kann, ein krankheitsrelevantes Protein seine Favoriten „wählen“ lassen und dann diese Protein‑Molekül‑Paare direkt wiegen kann, um zu sehen, welche am besten haften. Auf Galectin‑3 angewendet, fand diese Strategie unerwartet eine weniger erforschte Tasche und stärkte dort die Bindung, wodurch eine Verbindung entstand, die mit einigen der besten vorhandenen Inhibitoren konkurriert und als Lead für neue Krebstherapien dienen könnte. Allgemeiner bietet die Kopplung von in situ‑Chemie mit nativer Massenspektrometrie eine praktische Abkürzung zur Verfeinerung von Wirkstoff‑Leads gegen Proteine mit mehreren möglichen Bindungsstellen und kann Zeit, Material und Aufwand in den frühen Phasen der Wirkstoffentdeckung einsparen.
Zitation: Hoshi, K., Konuma, T., Taguchi, R. et al. Optimization of galectin-3 binding agents by in situ multiple compound synthesis and native mass spectrometry. Sci Rep 16, 8453 (2026). https://doi.org/10.1038/s41598-026-38570-z
Schlüsselwörter: Galectin‑3‑Inhibitoren, native Massenspektrometrie, fragmentbasierte Wirkstoffforschung, target‑guided synthesis, Krebsmedikament‑Leads