Clear Sky Science · de
Maschinelles Lern-Interatom-Potential für die strukturellen Eigenschaften von Eisenoxiden
Warum rostige Gesteine wichtig sind
Eisenoxide – die Minerale, die Rost seine Farbe verleihen – tragen stillschweigend wesentlich zum modernen Leben bei. Sie sind die Hauptquelle für Eisen zur Stahlherstellung, wichtige Bestandteile von Batterien und Solarzellen und helfen sogar bei der Reinigung verschmutzten Wassers. Dennoch fällt es uns trotz ihrer Bedeutung schwer, vorherzusagen, wie sich diese Materialien unter realen Bedingungen verhalten, insbesondere auf atomarer Ebene. Dieser Artikel beschreibt, wie Forschende moderne künstliche Intelligenz nutzten, um ein schnelles, präzises digitales Modell eines wichtigen Eisenoxids, des Hämatits, zu erstellen und damit zuverlässigere virtuelle Experimente von Erzverarbeitung bis hin zu Technologien für saubere Energie zu ermöglichen.
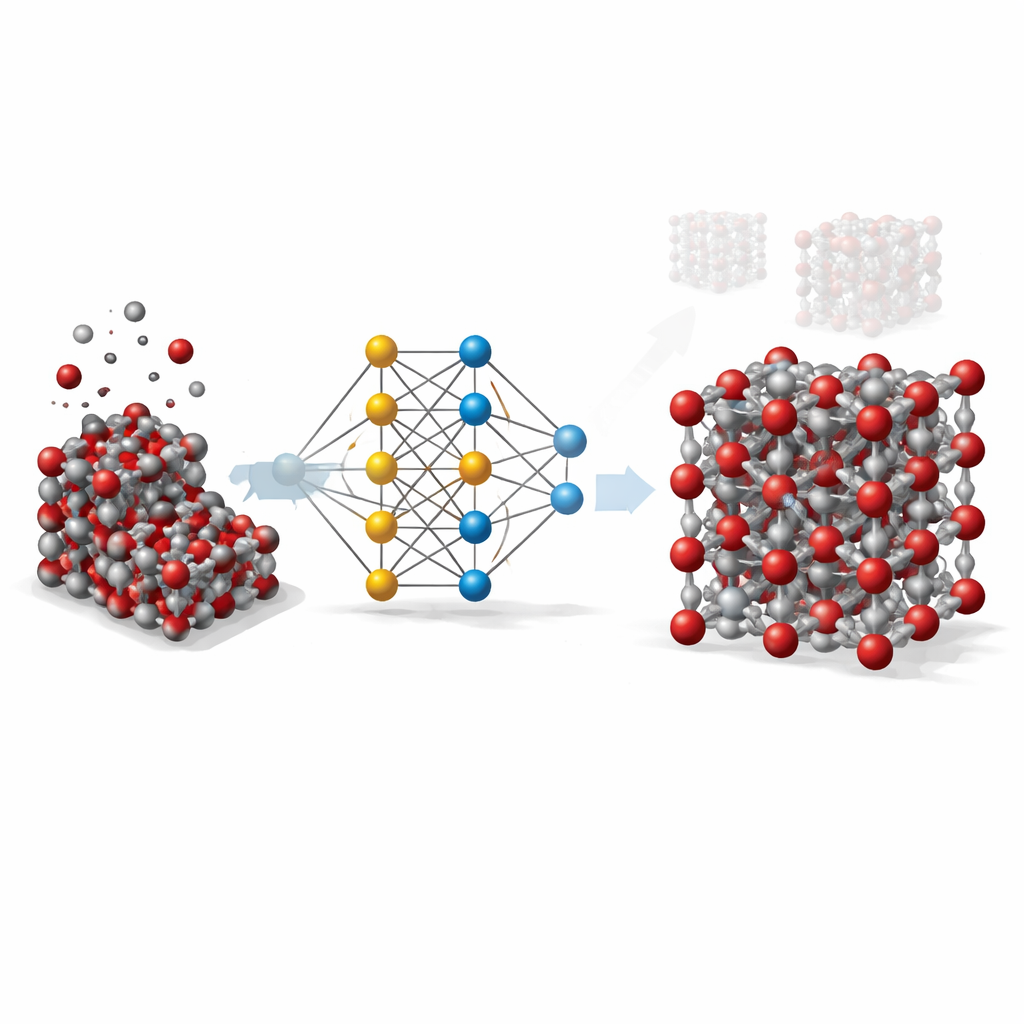
Von teuren Rechnungen zu schlauen Abkürzungen
Um einen Festkörper wie Hämatit im Detail zu verstehen, verwenden Wissenschaftler idealerweise quantenmechanische Methoden, die verfolgen, wie Elektronen und Atome miteinander wechselwirken. Diese Methoden sind zwar sehr genau, aber so rechenintensiv, dass sie für die Simulation großer Proben oder langer Zeiträume unpraktisch sind. Klassische Modelle sind dagegen schnell, aber grob: Sie beruhen auf einfachen Formeln, die für bestimmte Situationen angepasst wurden, und versagen oft, wenn Temperatur, Druck oder Kristallform sich ändern. Die hier vorgestellte Arbeit zielt darauf ab, diese Lücke zu schließen, indem maschinelles Lernen die Genauigkeit quantenmechanischer Berechnungen nachahmt und zugleich die Geschwindigkeit traditioneller Modelle beibehält.
Einem neuronalen Netz Atome beibringen
Das Team entwickelte ein sogenanntes Graph-Neural-Network-Potential für Hämatit. Bei diesem Ansatz wird jedes Atom als Knoten in einem Netzwerk behandelt, und Bindungen sowie Nachbaratome sind die Verbindungen zwischen den Knoten. Um dem Netzwerk beizubringen, wie Atome in Hämatit einander anziehen und abstoßen, erzeugten die Forschenden zunächst Tausende atomarer Momentaufnahmen mithilfe üblicher Simulationen über ein breites Spektrum an Temperaturen, Drücken und Kristalldeformationen, einschließlich sowohl Volumenkristallen als auch exponierten Oberflächen. Anschließend verwendeten sie eine hochwertige Quantenmethode (DFT+U), um Energie, Kräfte und innere Spannungen für jede Momentaufnahme zu berechnen, und trainierten das neuronale Netz darauf, diese Werte so genau wie möglich zu reproduzieren.
Das Modell an der Realität messen
Sobald das neue Potential – genannt Fe-MLIP – trainiert war, wurde es gründlich getestet. Die Autorinnen und Autoren verglichen seine Vorhersagen für grundlegende Strukturgrößen wie Gittermaße und die Verformung des Kristalls unter Belastung mit Experimenten und mehreren weitverbreiteten klassischen Modellen. Fe-MLIP gab die bekannte Kristallstruktur des Hämatits bis auf wenige Prozentpunkte korrekt wieder und erfasste sein elastisches Verhalten nahezu so gut wie direkte Quantenberechnungen, wodurch es viele klassische Kraftfelder bei zahlreichen Eigenschaften deutlich übertraf. Es erzielte auch gute Ergebnisse bei subtileren Tests, etwa wie sich das Material mit der Temperatur ausdehnt und wie seine Atome schwingen — Aspekte, die für Wärmeleitung und Spektroskopie wichtig sind. Diese Schwingungsfrequenzen, die während des Trainings nie explizit gezeigt wurden, lagen näher an gemessenen Werten als die der konkurrierenden Modelle.
Über ein einzelnes Mineral hinaus
Die Forschenden untersuchten anschließend, wie weit sich dieses auf Hämatit basierende Modell ausdehnen lässt. Sie wandten es auf verwandte Eisenoxide – Maghemit und Magnetit – an, die ähnliche atomare Bausteine teilen, sich aber in Kristallanordnung und Eisenladungszuständen unterscheiden. Obwohl Fe-MLIP nicht auf diesen Phasen trainiert wurde, lieferte es vernünftige Werte für deren Gittergrößen und Steifigkeit und erreichte dabei oft die Leistung oder übertraf spezialisierte klassische Modelle. Das Potential erfasste zudem die relative Stabilität wichtiger Kristalloberflächen und sogar Trends in den Energiekosten zur Erzeugung atomarer Vacanzen, Merkmale, die für das Verständnis von Korrosion, Katalyse und Batterieleistung entscheidend sind.
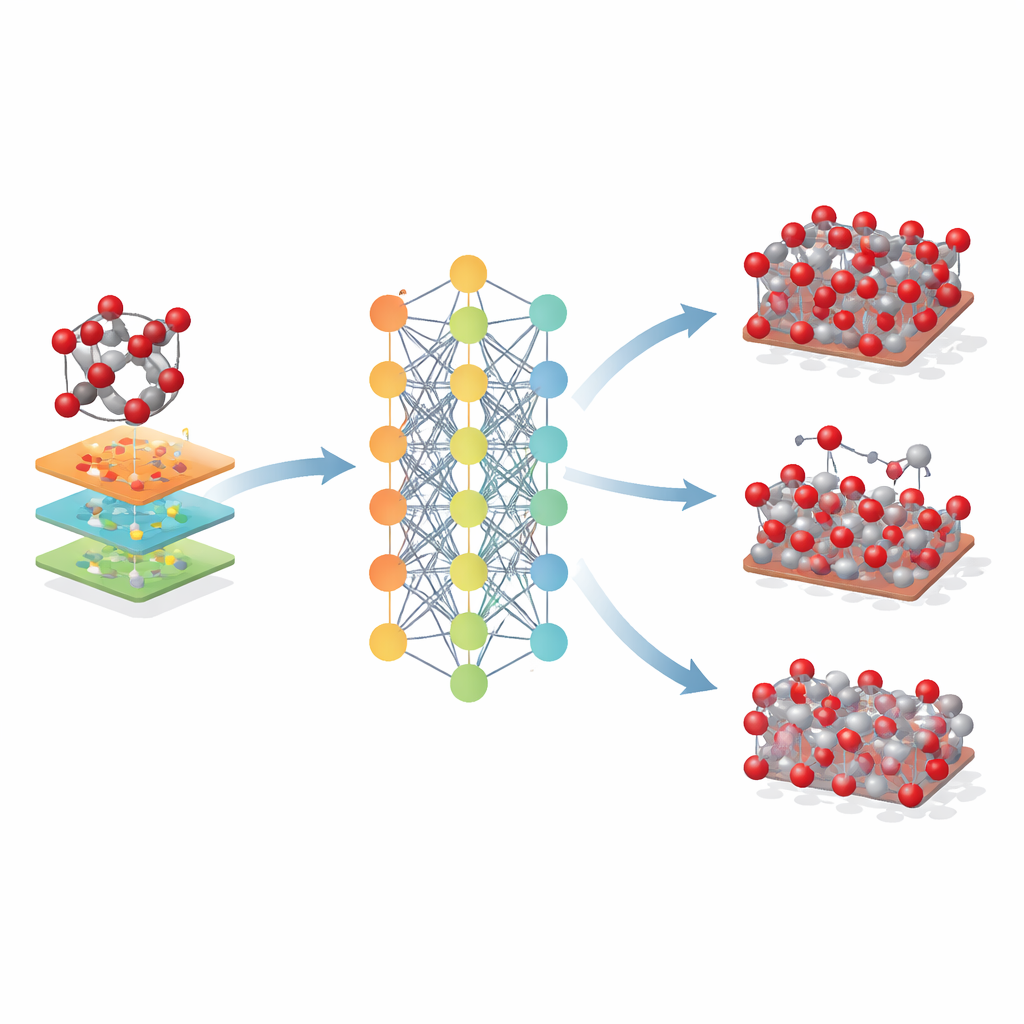
Was das für zukünftiges Materialdesign bedeutet
Für Nicht-Spezialisten lautet die Kernbotschaft, dass diese Arbeit einen leistungsfähigen neuen „digitalen Zwilling“ für Eisenoxide liefert. Das Fe-MLIP-Modell ermöglicht es Forschenden, große, lang dauernde Simulationen von Hämatit und verwandten Materialien mit nahezu quantenmechanischer Zuverlässigkeit, aber zu einem Bruchteil der Kosten durchzuführen. Zwar übernimmt es einige Beschränkungen der zugrunde liegenden Quantenmethode und konzentriert sich derzeit auf Eisen und Sauerstoff, doch erlaubt es bereits realistischere Studien darüber, wie diese Minerale auf Stress, Wärme, Oberflächen und Defekte reagieren. In praktischer Hinsicht kann ein solches Werkzeug die Entwicklung besserer Stahlherstellungsprozesse, effizienterer Katalysatoren und Batterien sowie verbesserter Umweltschutztechnologien, die auf Eisenoxiden basieren, beschleunigen — indem Wissenschaftler Ideen zuerst am Computer testen, bevor sie ins Labor oder ins Bergwerk gehen.
Zitation: Torres, A., de Oliveira, A.B., Barbosa, M.d.S. et al. Machine learning interatomic potential for the structural properties of iron oxides. Sci Rep 16, 8576 (2026). https://doi.org/10.1038/s41598-026-38096-4
Schlüsselwörter: Hämatit, Eisenoxide, maschinelles Lernpotenzial, Graph-Neuronale Netze, Molekulardynamik