Clear Sky Science · de
Molekulare Charakterisierung rezessiv vererbter ataktischer und neuropathischer Erkrankungen in konsanguinen pakistanischen Familien
Warum das für Familien und Ärzte wichtig ist
Störungen des Gleichgewichts, des Gehens und der Empfindung in Händen und Füßen können stark einschränkend sein, insbesondere wenn sie in der Kindheit beginnen und über das Leben langsam fortschreiten. Für viele Familien, vor allem in Regionen, in denen Cousin-Ehen häufig sind, treten diese Symptome über mehrere Generationen ohne klare Erklärung auf. Diese Studie geht eine drängende Frage für solche Familien in Pakistan an: Kann moderne DNA-Analyse endlich die verborgenen genetischen Ursachen ihrer Ataxie (gestörtes Gleichgewicht und Koordination) und peripheren Neuropathie (Schädigung der Nerven in den Gliedmaßen) aufdecken und Ärzten helfen, klarere Diagnosen und mögliche Behandlungsansätze anzubieten?
Verfolgen vererbter Erkrankungen in großen Familien
Die Forschenden arbeiteten mit sieben weit verzweigten pakistanischen Familien, in denen mehrere Angehörige schwere Bewegungs- und Nervenprobleme hatten. Bei einigen standen Ataxiesymptome im Vordergrund, die das sichere Gehen oder die Kontrolle von Sprache und Augenbewegungen erschwerten. Andere zeigten typische Zeichen peripherer Neuropathie, wie Muskelschwund in Händen und Füßen, Fußdeformitäten und Reflexverlust. In diesen Familien waren die Eltern miteinander verwandt, was die Wahrscheinlichkeit erhöht, dass Kinder zwei Kopien desselben seltenen fehlerhaften Gens erben. Mit Blutproben von betroffenen und nicht betroffenen Verwandten führten die Forschenden Exom-Sequenzierung durch — das Lesen nahezu aller protein-kodierenden Teile des Genoms —, um nach schädlichen Veränderungen zu suchen, die sich über die Familienstammbäume mit der Erkrankung verfolgen ließen. 
Lokalisierung seltener schädlicher Gene
Durch das Herausfiltern häufiger und harmloser DNA-Varianten identifizierten die Wissenschaftler in fünf der sieben Familien wahrscheinlich krankheitsverursachende Varianten. Jede dieser Familien trug ihre eigene spezifische genetische Veränderung, und alle zeigten ein rezessives Vererbungsmuster: Krank wurden Menschen nur, wenn sie zwei fehlerhafte Kopien, eine von jedem Elternteil, erbten. In einer Familie mit im Erwachsenenalter auftretenden Gleichgewichtsproblemen und Sprachstörungen war die Ursache eine seltene Veränderung im Gen MFSD8, das beim Transport von Materialien in zelluläre Recycling-Kompartimente, die Lysosomen, hilft. In einer anderen Familie stand eine schädigende Veränderung in AFG3L2, einem Protein, das die Gesundheit der Mitochondrien — den Kraftwerken der Zelle — erhält, in Verbindung mit einer im Kindesalter beginnenden spastischen Ataxie mit Dystonie (abnorme Muskelkontraktionen). Eine dritte Familie trug einen Frameshift-Fehler in SETX, einem Gen, das die DNA während der Reparatur schützt und bereits dafür bekannt ist, Ataxie mit okulomotorischer Apraxie zu verursachen, eine Erkrankung, die ebenfalls die Augenbewegungen betrifft.
Ein näherer Blick auf vererbten Nervenschaden
Zwei weitere Familien hatten eine Form der Charcot-Marie-Tooth-(CMT)-Erkrankung, einer Gruppe erblich bedingter Krankheiten, die die langen Nerven zu Füßen und Händen schädigen. In beiden Fällen fanden die Forschenden schädigende Varianten im Gen GDAP1, das für die normale mitochondriale Funktion in Nervenzellen entscheidend ist. Eine GDAP1-Veränderung verkürzte das Protein stark und war mit einer sehr schweren, früh beginnenden Erkrankung assoziiert; eine andere tauschte eine einzelne Aminosäure im Protein aus und führte zu einem etwas milderen Verlauf. Auffällig war, dass der schwerstbetroffene Patient in einer CMT-Familie zudem homozygot für eine bekannte krankheitsverursachende Variante in einem zweiten Gen, MMACHC, war. MMACHC ist an der Verwertung von Vitamin B12 beteiligt und kann teilweise mit vitaminbasierten Therapien behandelbar sein. Dieser doppelte Befall könnte erklären, warum seine Symptome ausgeprägter waren als die seiner Verwandten ohne die MMACHC-Variante.
Wenn die DNA-Suche nicht ausreicht
Nicht jede Familie lieferte eine klare genetische Erklärung. In zwei der sieben Familien konnte das Team keine einzelne Veränderung im Exom finden, die überzeugend zum Erkrankungsmuster passte. In einem Fall identifizierten sie eine Variante im Gen EHHADH, die zwar dem Vererbungsmuster entsprach, aber als harmlos vorhergesagt wird und bekannt dafür ist, bei Veränderungen eine andere, nierenbezogene Erkrankung zu verursachen. In einem anderen Fall stellten sich zwei Cousins mit ähnlichen Bewegungsstörungen als unterschiedlich assoziiert heraus: Ein Junge trug eine bekannte schädliche Variante in ALS2, die zu jugendlichen Formen der Motoneuronerkrankung führen kann, während seine betroffenen Cousins diese Variante nicht hatten. Diese ungelösten Fälle deuten darauf hin, dass wichtige Mutationen in Regionen des Genoms liegen könnten, die die Standard-Exom-Sequenzierung nicht erfasst, oder dass mehr als ein subtiler genetischer Faktor miteinander interagiert.
Was das für Patienten und künftige Versorgung bedeutet
Insgesamt zeigen die Ergebnisse, dass leistungsfähige DNA-Werkzeuge die spezifischen Gene hinter komplexen Nerven- und Gleichgewichtsstörungen aufdecken können, selbst in ressourcenbegrenzten Umgebungen. Für die fünf Familien mit eindeutigen Befunden verwandelt die Arbeit vage Bezeichnungen wie „Ataxie" oder „Neuropathie" in präzise Diagnosen, die an bestimmte Gene gebunden sind; das kann die genetische Beratung leiten, die Prognose informieren und in manchen Fällen Behandlungsmöglichkeiten aufzeigen, etwa vitamin-B12-bezogene Therapien bei MMACHC-assoziierter Erkrankung. Die Studie erweitert zudem das wissenschaftliche Verständnis darüber, wie Gene wie MFSD8, AFG3L2, SETX, GDAP1, MMACHC und ALS2 die Gesundheit von Nervenzellen im Gehirn, Rückenmark und peripheren Nerven beeinflussen. Für die Zukunft werden umfassendere Genomsequenzierung und funktionelle Studien nötig sein, um die verbleibenden Rätsel zu lösen und diese genetischen Erkenntnisse in frühere Diagnosen und bessere Versorgung für betroffene Kinder und Erwachsene zu überführen. 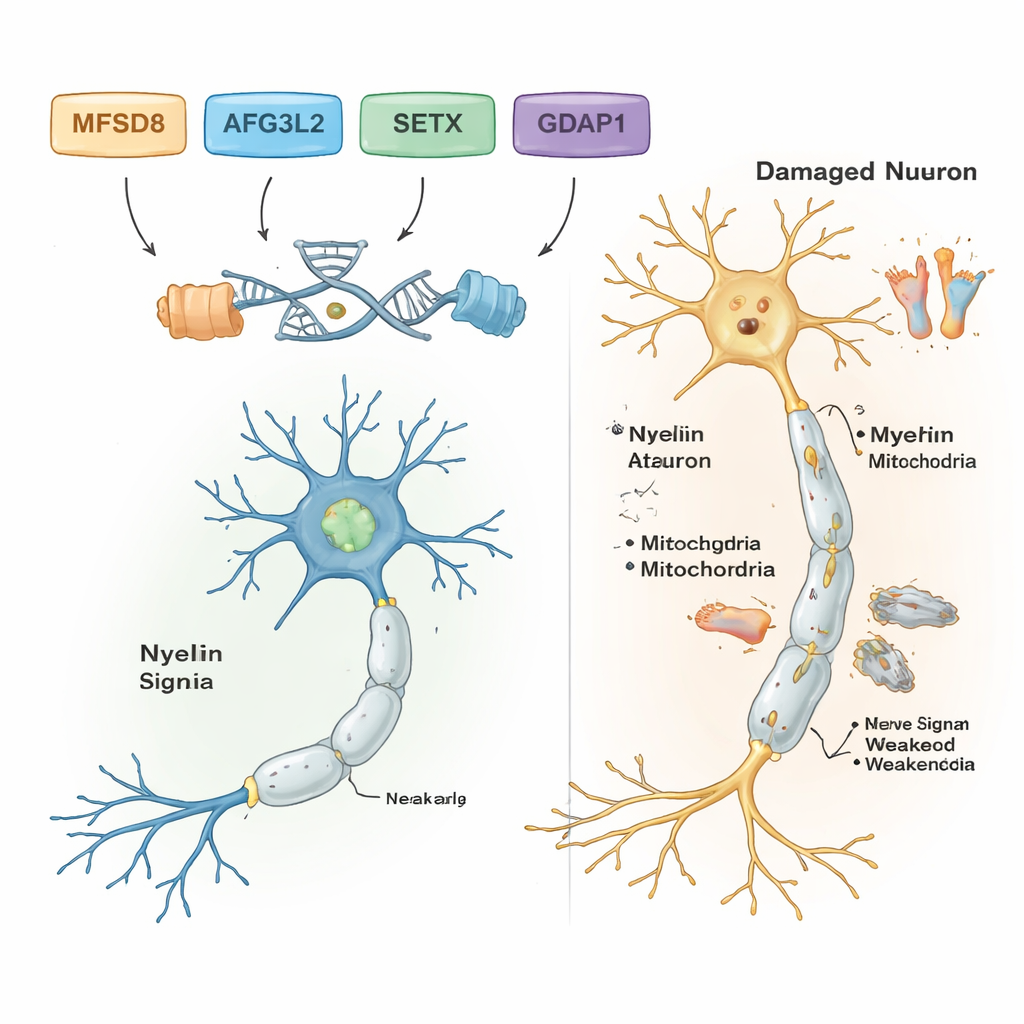
Zitation: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Schlüsselwörter: Ataxie, periphere Neuropathie, Exom-Sequenzierung, Charcot-Marie-Tooth-Krankheit, genetische Diagnose