Clear Sky Science · de
Optimierung der Merkmalsauswahl in Krebs‑Microarray‑Daten mittels eines heap‑gesteuerten evolutionären Frameworks für hochdimensionale Räume
Warum die richtige Genwahl wichtig ist
Moderne Gentests können Zehntausende von Genen gleichzeitig messen, während Ärztinnen und Ärzte oft nur Daten von einigen Dutzend Patientinnen und Patienten haben. Versteckt in diesem riesigen „Gen‑Dschungel“ befinden sich deutlich weniger Signale, die wirklich eine Krebsart von einer anderen oder Tumorgewebe von gesundem Gewebe unterscheiden. Dieses Papier stellt eine neue Suchstrategie vor, die automatisch diese Schlüsselsegmente identifiziert, mit dem Ziel, computerunterstützte Krebsdiagnosen genauer, schneller und besser interpretierbar zu machen.
Zu viele Signale, zu wenige Daten
Microarray‑Experimente und ähnliche Technologien erlauben es, in jeder Probe Aktivitätsniveaus für Tausende von Genen zu erfassen. Die Anzahl der Proben ist jedoch meist sehr gering, mitunter unter hundert. Viele dieser Genmessungen sind verrauscht, redundant oder für die betrachtete Erkrankung irrelevant. Alle beizubehalten kann Lernalgorithmen überfordern, Berechnungen verlangsamen und zu irreführenden Modellen führen, die sich an zufälligen Auffälligkeiten statt an echter Biologie festhalten. Der Prozess, dies auf eine nützliche Teilmenge zu reduzieren, heißt „Merkmalsauswahl“ und ist entscheidend, wenn man verlässliche Vorhersagen aus hochdimensionalen medizinischen Daten erhalten möchte.
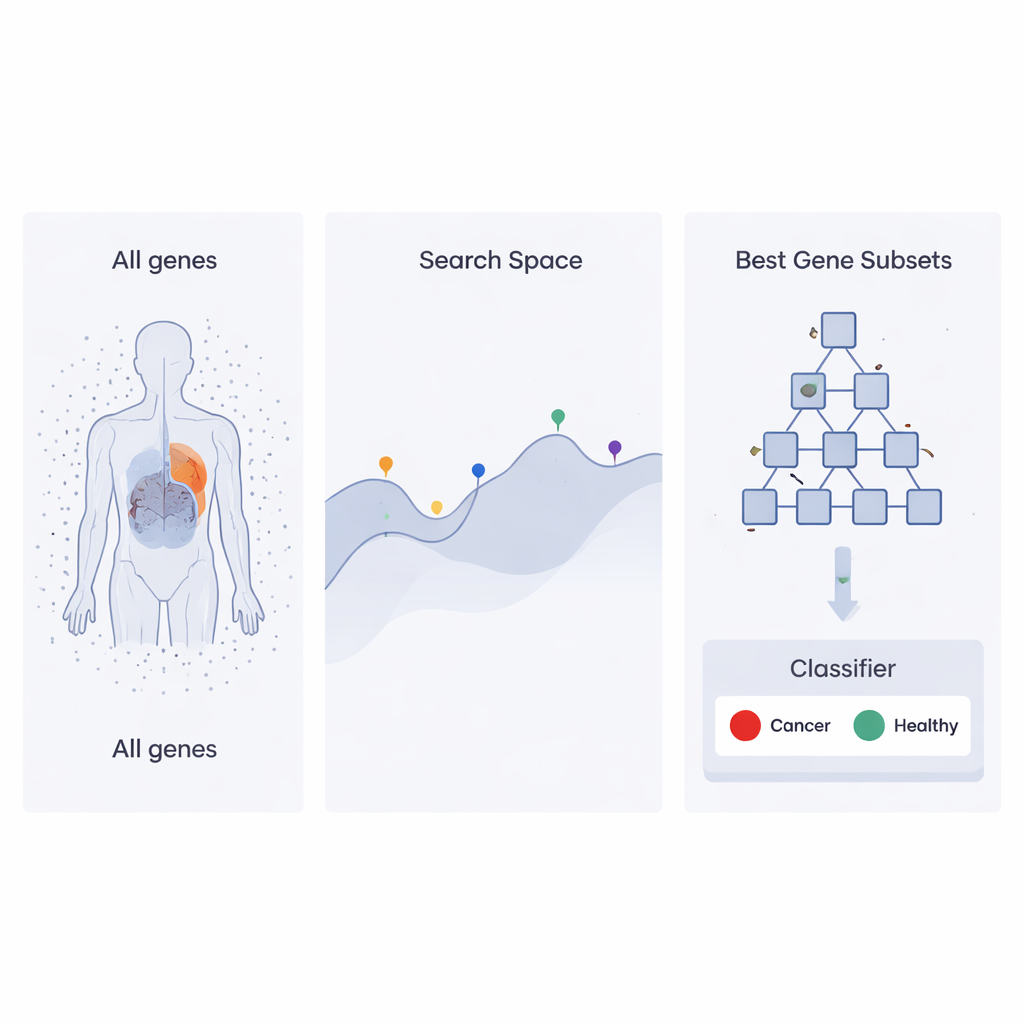
Eine Suchstrategie, inspiriert von Firmenhierarchien
Die Autorinnen und Autoren bauen auf einem aktuellen Optimierungsansatz auf, dem Heap‑Based Optimizer (HBO), der Ideen aus der Mitarbeiterorganisation in Unternehmen übernimmt. Stellen Sie sich jede mögliche Genmenge als „Mitarbeiter“ vor, dessen Leistung danach beurteilt wird, wie gut sie einem Klassifikator hilft, Krebsproben von gesunden zu unterscheiden. Diese „Mitarbeiter“ werden in einer Hierarchie angeordnet, ähnlich einer Firmenleiter, mithilfe einer Computerstruktur, die als Heap bekannt ist. Leistungsstarke Genmengen sitzen weit oben, schwächere weiter unten. In vielen Durchläufen passen niedrig eingestufte „Mitarbeiter“ ihre Auswahl an, indem sie von Vorgesetzten und Kollegen kopieren und dies leicht modifizieren, wodurch die gesamte Organisation schrittweise bessere Lösungen findet.
Rohdaten in schärfere Muster verwandeln
Um die Suche effektiver zu machen, verlassen sich die Autorinnen und Autoren nicht ausschließlich auf rohe Genwerte. Zuerst formen sie die Microarray‑Daten in eine bildähnliche Struktur und wenden eine Technik namens Histogram of Oriented Gradients (HOG) an, die in der Computer Vision weit verbreitet ist. HOG erfasst, wie sich Expressionsniveaus über Gene hinweg verändern und hebt lokale Muster hervor, statt isolierte Messwerte. Diese musterbasierten Merkmale werden dann mit den ursprünglichen Geninformationen kombiniert. Ein einfacher Klassifikator, k‑Nearest Neighbors (KNN), fungiert als „Richter“ und bewertet jede Kandidaten‑Genmenge nach ihrer Genauigkeit bei der Kennzeichnung neuer Proben, wobei außerdem kompaktere Mengen belohnt werden.
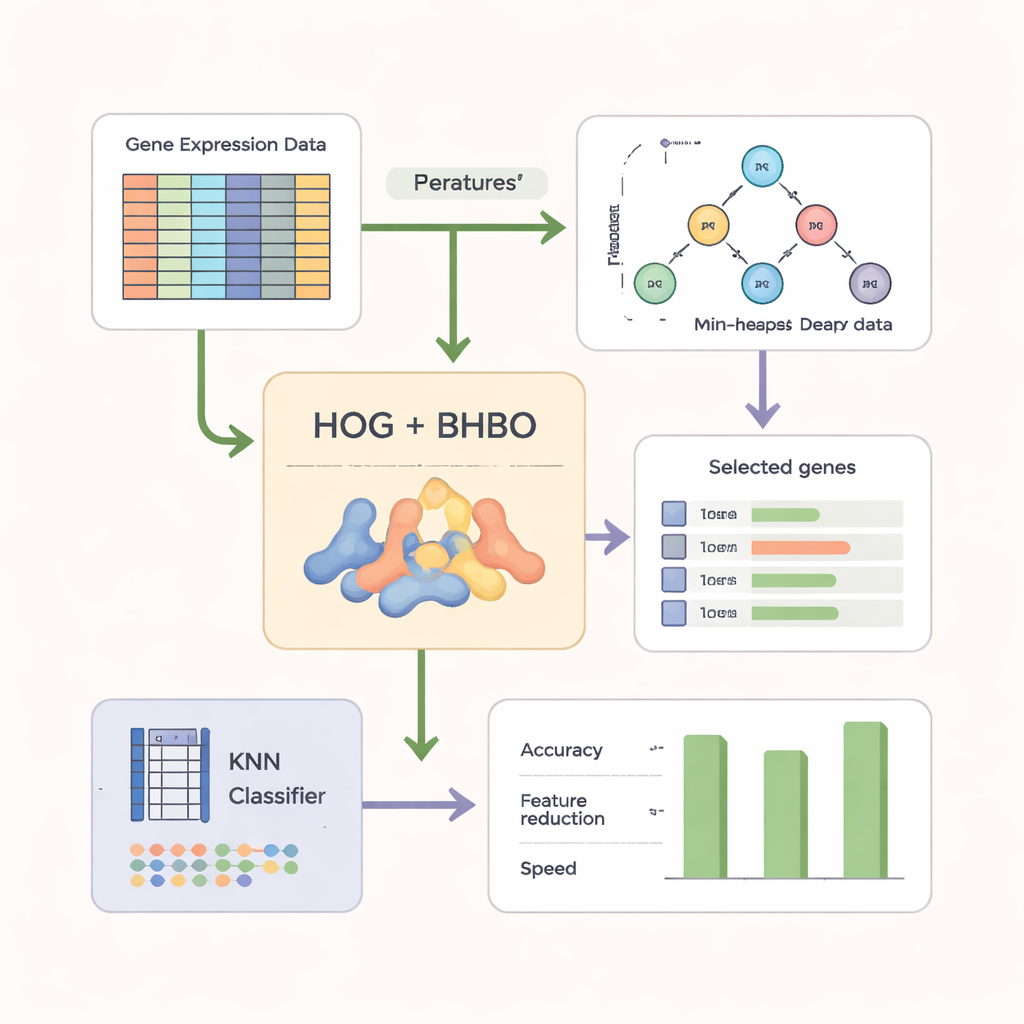
Test auf mehreren Krebsdatensätzen
Die Forschenden bewerteten ihre binäre Variante des Heap‑Based Optimizer (BHBO) an neun öffentlichen Krebs‑Microarray‑Datensätzen, darunter Gehirntumore, Leukämien, Prostatakrebs und gemischte Tumorsammlungen mit vielen Subtypen. Jeder Datensatz enthielt Tausende bis über fünfzehntausend gemessene Gene, aber relativ wenige Proben. Für jeden Datensatz wurde BHBO wiederholt ausgeführt und mit sieben bekannten Suchmethoden verglichen, etwa genetischen Algorithmen und Partikelschwarmoptimierung. Gemessen wurden nicht nur die Genauigkeit, sondern auch die Anzahl der behaltenen Gene, die Geschwindigkeit der Konvergenz und die Stabilität der Ergebnisse bei gestörten Daten durch simuliertes Rauschen, Batch‑Effekte und falsche Labels.
Was die neue Methode erreichte
Über alle neun Datensätze hinweg erreichte der heap‑gesteuerte Ansatz eine durchschnittliche Klassifikationsgenauigkeit von rund 95 Prozent und reduzierte die Anzahl der Gene um mehr als 85 Prozent. In mehreren Datensätzen übertraf er deutlich konkurrierende Methoden und zeigte eine schnellere Konvergenz, das heißt er fand in weniger Suchschritten gute Genmengen. Selbst wenn die Autorinnen und Autoren die Daten absichtlich verfälschten – durch Hinzufügen von Rauschen oder Umdrehen einiger Probenlabels – sank die Leistung nur leicht und blieb besser als die Alternativen. Statistische Tests bestätigten, dass diese Verbesserungen kaum zufällig sind.
Was das für zukünftige Krebsdiagnostik bedeutet
Praktisch zeigt diese Arbeit, dass eine sorgfältig gestaltete Suchstrategie riesige genetische Datensätze durchforsten und kleine, informationsreiche Genpanels entdecken kann, die Krebs weiterhin sehr gut klassifizieren. Für Klinik und Forschung sind solche kompakten Genmengen leichter biologisch zu validieren, kostengünstiger in Folgeuntersuchungen zu messen und besser für die Integration in Entscheidungsunterstützungssysteme geeignet. Obwohl die Methode nicht direkt neue Medikamente oder Signalwege entdeckt, richtet sie den Fokus stärker auf vielversprechende genetische Marker und hilft anderen Studien, sich auf die aussagekräftigsten Signale in hochdimensionalen Krebsdaten zu konzentrieren.
Zitation: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Schlüsselwörter: Krebs‑Microarray, Merkmalsauswahl, Metaheuristische Optimierung, Gen‑Biomarker, Medizinisches Data‑Mining