Clear Sky Science · de
Eine Methode zur Erkennung struktureller Varianten mit Hi-C-Kontaktmatrix und neuronalen Netzen
Warum das Biegen von DNA im 3D wichtig ist
Unsere DNA wird oft als einfache Buchstabenreihe dargestellt, faltet sich in jeder Zelle jedoch zu einer komplexen dreidimensionalen Struktur. Wenn große Abschnitte dieser Reihe gelöscht, umgedreht oder verschoben werden – Veränderungen, die als strukturelle Varianten bezeichnet werden –, können sie Gene stören und Krebs fördern. Diese Studie stellt VarHiCNet vor, ein neues künstliches Intelligenzsystem, das 3D-Kartierungen der DNA faltung ausliest und solche riskanten großräumigen Veränderungen genauer erkennt als bestehende Werkzeuge. Das bietet einen neuen Ansatz zur Untersuchung von Krebsgenomen und anderen Krankheiten.
Genomveränderungen durch 3D-Kontaktkarten sehen
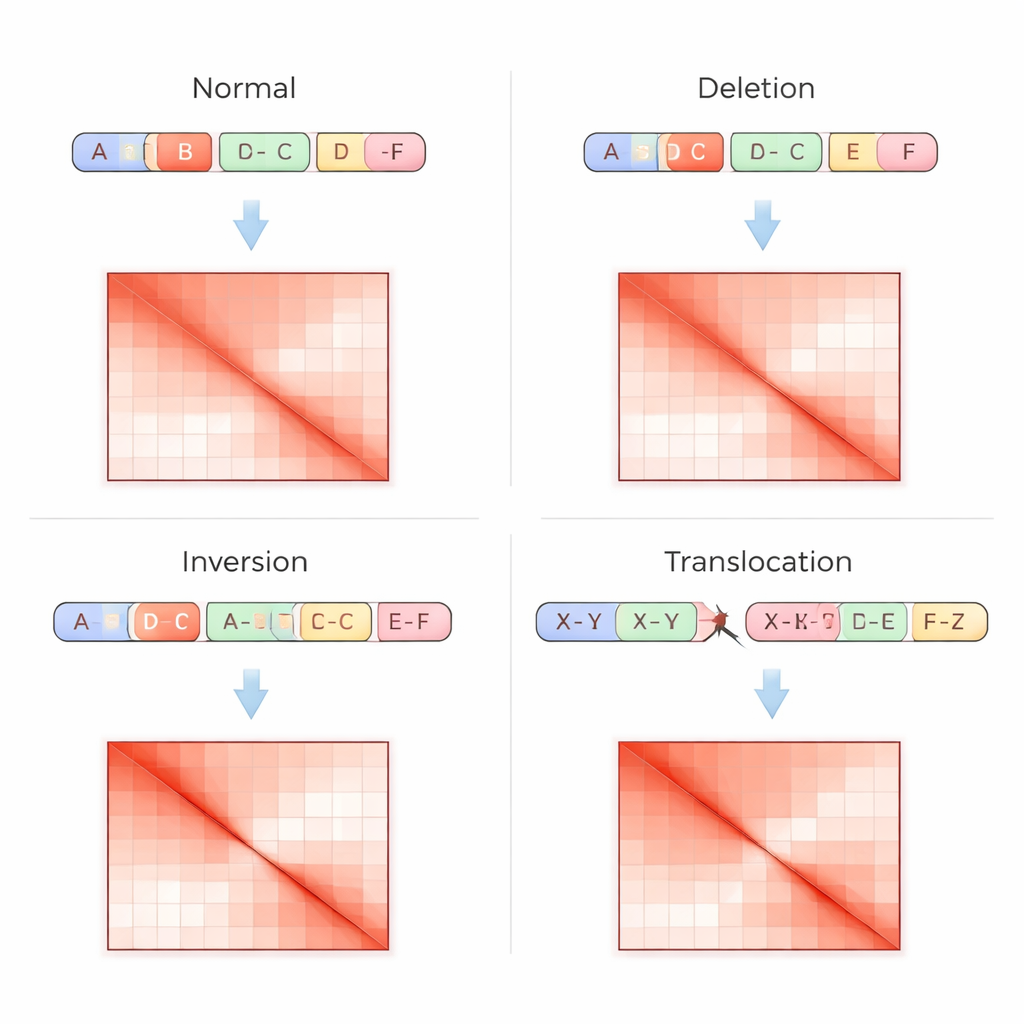
Traditionelle Genomtests lesen DNA als lineare Sequenz, was das Erkennen komplexer Umlagerungen erschwert, besonders in repetitiven Regionen oder wenn Abschnitte verschoben werden, ohne ihre Kopienzahl zu ändern. Die Hi-C-Technik geht das Problem anders an: Sie misst, wie oft weit entfernte DNA-Teile im Zellkern physisch in Kontakt kommen, und zeichnet diese Kontakte als Raster oder Kontaktmatrix auf, wobei hellere Stellen stärkere Interaktionen bedeuten. Strukturelle Varianten hinterlassen charakteristische Fingerabdrücke in diesen Matrizen – wie fehlende Streifen, wenn ein Bereich gelöscht wurde, gespiegelte Muster bei einer Inversion oder außendiagonale Hotspots, wenn zwei Chromosomen miteinander verschmolzen sind. VarHiCNet ist darauf ausgelegt, diese visuellen Muster automatisch zu erkennen.
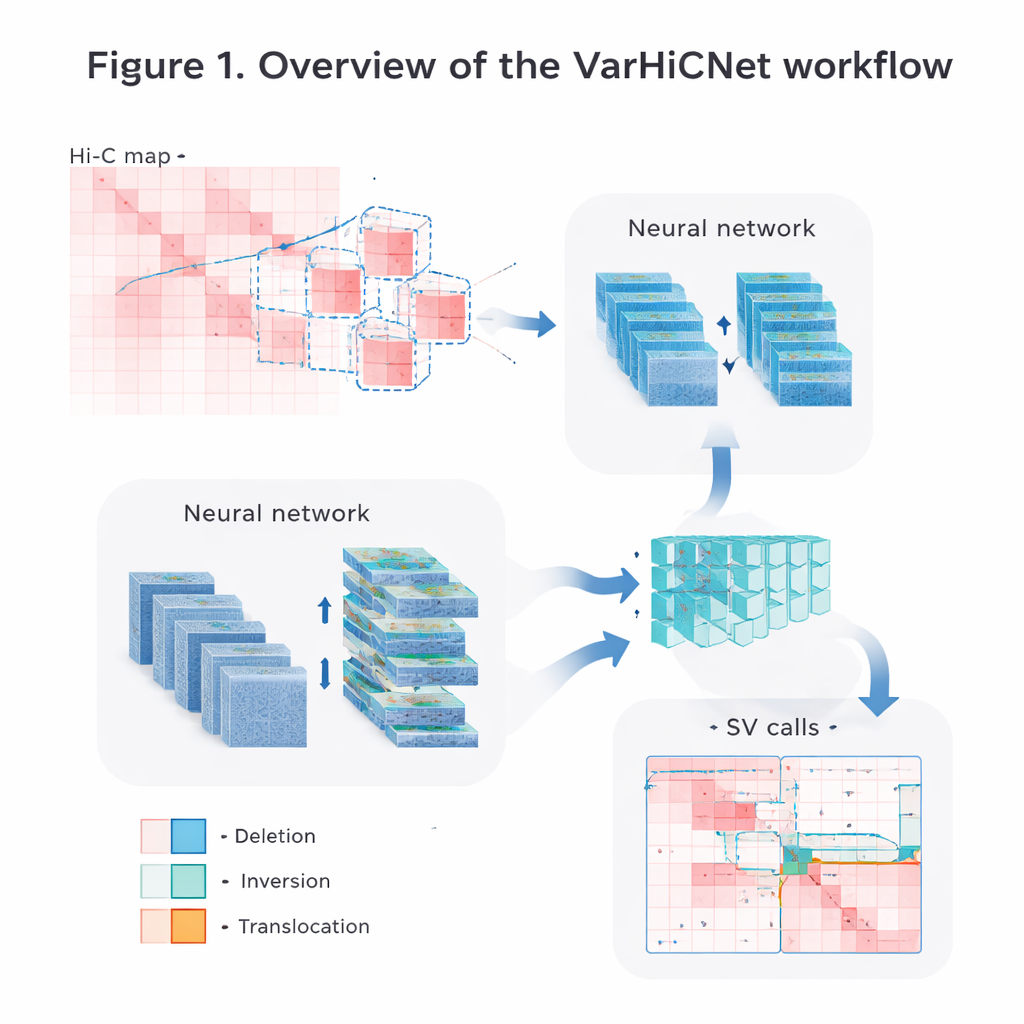
Genomkarten in Bilder für KI verwandeln
Die Autoren wandeln die rohen Hi-C-Kontaktdaten in Bilder um, die von Computer-Vision-Systemen leicht verarbeitet werden können. Zuerst normalisieren sie die Matrizen sorgfältig, um den natürlichen Abfall der Kontaktfrequenz mit zunehmender Distanz zu korrigieren, wobei sowohl nahe als auch fernere Interaktionssignale erhalten bleiben. Dann scannen sie jedes Chromosom mit sich überlappenden quadratischen Fenstern und schneiden viele kleinere Untermatrizen aus. Jede Untermatrix wird in ein standardisiertes 800×800-Pixel-Farbbild skaliert, wobei unterschiedliche Kontaktstärken in rötlichen Intensitäten über drei Farbkanäle abgebildet werden. Diese bildähnliche Darstellung erlaubt dem Modell, leistungsfähige Techniken wiederzuverwenden, die ursprünglich für die Erkennung von Objekten in Fotografien entwickelt wurden.
Tricks aus der Objekterkennung übernehmen
VarHiCNet behandelt jede potenzielle strukturelle Variante wie ein „Objekt“ in einem Bild. Es baut auf einem modernen Objekterkennungs-Framework namens RT-DETR auf, das eine Kombination aus Convolutional Neural Networks und Transformern nutzt, um wichtige Regionen hervorzuheben. Ein ResNet-Backbone extrahiert zunächst Merkmale auf mehreren Skalen: flache Schichten bewahren feine Details, die nötig sind, um exakte Bruchpunkte zu lokalisieren, während tiefere Schichten breitere Muster erfassen, die auf große Ereignisse hinweisen. Ein Feature-Fusion-Modul mischt dann Informationen aus mehreren Schichten, sodass sowohl lokale als auch globale Hinweise erhalten bleiben. Ein weiterer kundenspezifischer Baustein, inspiriert von Spatial Pyramid Pooling, passt an, wie viel der Umgebung das Modell gleichzeitig „sieht“, wodurch es empfindlich wird für Varianten, die von relativ kleinen bis hin zu sehr großen DNA-Abschnitten reichen.
Von Kandidatenregionen zu präzisen Variantentypen
Sobald VarHiCNet Kandidatenregionen im Hi-C-Bild vorgeschlagen hat, muss es diese zu exakten Bruchpunkten und spezifischen Variantentypen verfeinern, wie Deletionen, Inversionen, Duplikationen oder Translokationen. Dazu zoome das System in die Umgebung jedes vorhergesagten Bruchpunkts und reduziere deren Komplexität mittels einer mathematischen Technik namens Hauptkomponentenanalyse, die hervorhebt, wo sich das Kontaktmuster am stärksten ändert. Diese kompakten Repräsentationen werden dann in einen Transformer-basierten Klassifikator eingespeist, der feine Unterschiede in den lokalen Mustern für jede Variantenkategorie erlernt. Das Ergebnis ist ein detaillierter Aufruf für jedes Ereignis: wo es im Genom auftritt und welche Art struktureller Veränderung es darstellt.
Leistung über verschiedene Krebszelllinien hinweg
Die Forschenden testeten VarHiCNet an Hi-C-Daten aus sechs verschiedenen menschlichen Krebszelllinien, die Blut-, Brust-, Hirn-, Nieren-, Lungen- und Prostatatumore abdecken. Mithilfe eines hochvertrauenswürdigen Katalogs bekannter struktureller Varianten als Goldstandard verglichen sie ihre Methode mit mehreren führenden Tools, die ebenfalls Hi-C-Daten analysieren. Sowohl bei intrachromosomalen als auch bei interchromosomalen Ereignissen erzielte VarHiCNet im Allgemeinen höhere oder vergleichbare F1-Werte, was bedeutet, dass es Sensitivität und Genauigkeit besser ausbalanciert als andere Ansätze. Besonders stark war es beim Erkennen balancierter Translokationen und Inversionen – Umlagerungen, die in konventionellen DNA-Sequenzierungen oft kaum Spuren hinterlassen, in 3D-Faltungssignaturen jedoch deutlich sichtbar sind. Die Autoren zeigten außerdem, dass ihre Designentscheidungen, etwa die Bildauflösung und die Feature-Fusion-Module, die Leistung in kontrollierten Tests konsequent verbesserten.
Was das für das Verständnis von Krankheiten bedeutet
Vereinfacht gesagt bietet VarHiCNet Wissenschaftlern eine intelligentere Möglichkeit, darauf zu „blicken“, wie das Genom im 3D gefaltet ist, und großräumige krankheitsrelevante Umlagerungen zu erkennen, die durch konventionelle Sequenzierung allein übersehen werden könnten. Indem komplexe Kontaktkarten in Bilder verwandelt und moderne, an die Bildverarbeitung angelehnte neuronale Netze angewendet werden, kann die Methode viele Arten struktureller Varianten mit hoher Zuverlässigkeit über verschiedene Krebszelltypen hinweg erkennen und kategorisieren. Obwohl sie bei sehr kleinen oder stark verwobenen Veränderungen noch Schwierigkeiten hat und auf umfangreiche Trainingsdaten angewiesen ist, deutet VarHiCNet auf eine Zukunft hin, in der die 3D-Genomarchitektur routinemäßig Teil dessen wird, wie wir genetische Veränderungen lesen, interpretieren und schließlich gezielt gegen Krebs und andere Krankheiten vorgehen.
Zitation: Shen, J., Wang, H., Zhai, H. et al. A method for structural variant detection using Hi-C contact matrix and neural networks. Sci Rep 16, 7324 (2026). https://doi.org/10.1038/s41598-026-37678-6
Schlüsselwörter: strukturelle Variation, Hi-C, Deep Learning, Krebsgenomik, 3D-Genom