Clear Sky Science · de
Ein DNABERT-basiertes Deep-Learning-Framework zur Vorhersage von Transkriptionsfaktor-Bindungsstellen
Warum die Vorhersage von DNA-Steuerungen wichtig ist
Jede Zelle in Ihrem Körper trägt im Wesentlichen dieselbe DNA, dennoch verhalten sich Nervenzellen, Leberzellen und Immunzellen sehr unterschiedlich. Ein Grund dafür sind spezielle Proteine, sogenannte Transkriptionsfaktoren, die wie molekulare Schalter wirken und Gene ein- oder ausschalten, indem sie an kurze DNA-Abschnitte andocken, die man Bindungsstellen nennt. Diese Andockstellen experimentell im ganzen Genom zu finden, ist zeitaufwendig und teuer. Diese Studie stellt TFBS-Finder vor, ein neues KI-Modell, das rohe DNA-Buchstaben liest und genauer vorhersagen kann, wo Transkriptionsfaktoren binden — was die Forschung zu Genregulation und Krankheit möglicherweise beschleunigen kann.
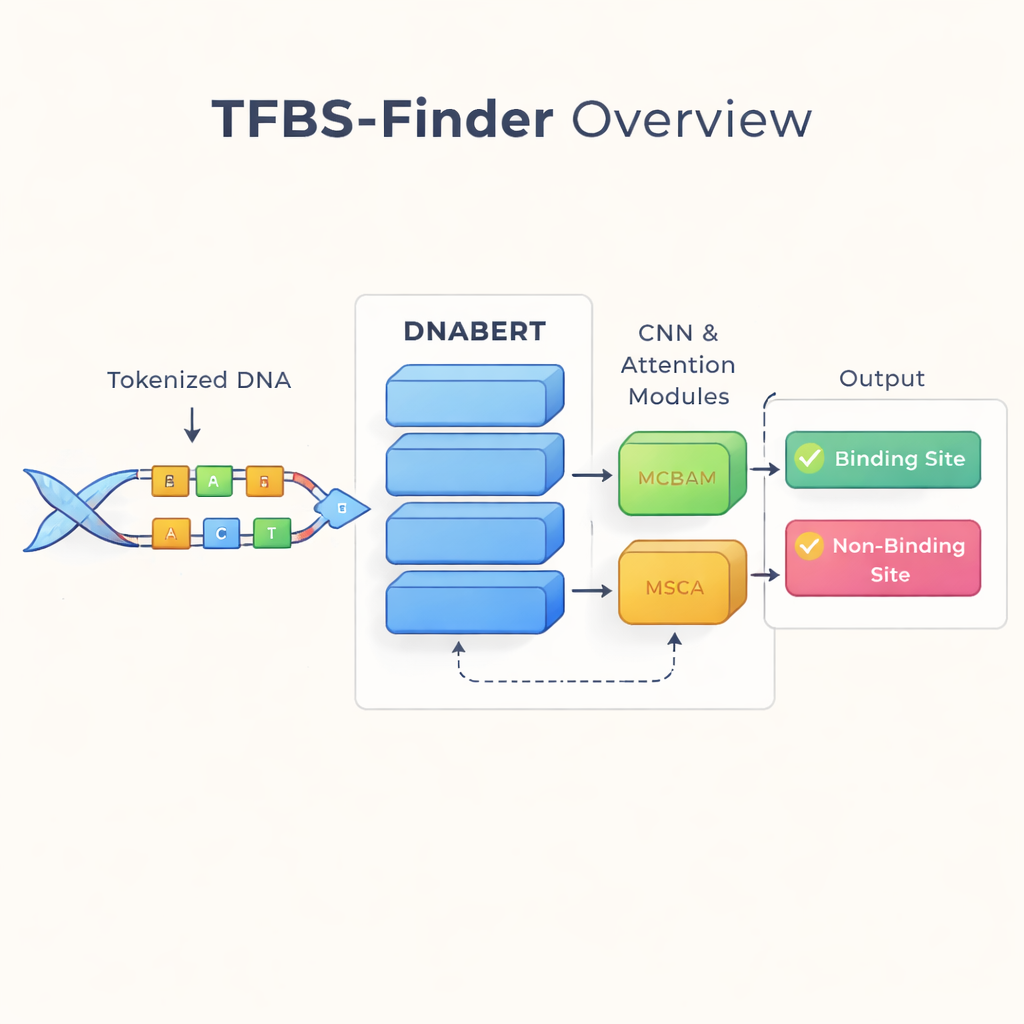
DNA wie eine Sprache lesen
Die Autoren bauen auf einer Idee auf, die die Sprachtechnologie verändert hat: Behandle DNA, als wäre sie Text. Sie verwenden DNABERT, eine Version des BERT-Sprachmodells, die statt auf Wörtern auf menschlicher DNA nachtrainiert wurde. DNABERT betrachtet nicht nur einzelne Buchstaben; es zerlegt die DNA in sich überlappende kurze „Wörter“ aus fünf Buchstaben und lernt, wie diese Stücke gemeinsam auftreten. So kann das Modell Kontext über weite Strecken erfassen, beispielsweise wie Muster an einem Ende einer Sequenz mit Mustern weit entfernt zusammenhängen — ähnlich wie das Verstehen der Bedeutung eines Satzes statt isolierter Wörter.
Lokale Muster mit fokussierter Aufmerksamkeit finden
Während DNABERT gut darin ist, den globalen Kontext zu erfassen, hängen Transkriptionsfaktor-Bindungen oft von sehr kurzen, präzisen Motiven ab — lokalen Mustern in der DNA. TFBS-Finder ergänzt DNABERT daher um mehrere zusätzliche Komponenten. Ein Convolutional Neural Network (CNN) durchkämmt die Sequenzeinbettungen, um wiederkehrende lokale Formen hervorzuheben, ähnlich wie Bildverarbeitungssoftware Kanten und Ecken erkennt. Zwei Attention-Module, MCBAM und MSCA genannt, wirken dann wie einstellbare Scheinwerfer, die die informativsten Merkmale verstärken und Rauschen abschwächen. Zusammen balancieren diese Bausteine den Gesamtzusammenhang mit feingliedrigen Details aus, um zu entscheiden, ob ein DNA-Abschnitt eine echte Bindungsstelle enthält.
Beweisen, dass jedes Bauteil wirklich hilft
Um zu prüfen, ob all diese Komponenten notwendig sind, führte das Team umfangreiche Ablations-Experimente durch, bei denen Module systematisch entfernt oder umgestellt und das System neu auf 165 Benchmark-Datensätzen trainiert wurde, die 29 Transkriptionsfaktoren über 32 Zelltypen abdecken. Anhand gängiger Bewertungsmaße für Vorhersagequalität belegte das vollständige TFBS-Finder-Modell durchgängig Spitzenwerte. Einfachere Varianten, die nur DNABERT nutzten oder eines der Attention-Module wegließen, verloren deutlich an Genauigkeit. Statistische Tests bestätigten, dass diese Leistungsabfälle nicht zufällig waren und zeigten, dass die Kombination aus globalem Sequenzverständnis und sorgfältig gestalteter Aufmerksamkeit für lokale Muster entscheidend ist.
Über Zelltypen hinweg arbeiten und ältere Werkzeuge übertreffen
Eine wichtige Frage ist, ob ein in einem biologischen Kontext trainiertes Modell auf einen anderen generalisieren kann. Die Autoren konzentrierten sich auf einen gut untersuchten Transkriptionsfaktor namens CTCF und trainierten TFBS-Finder mit Daten einer Zelllinie, um es dann in anderen zu testen. In allen Kombinationen erreichte das Modell hohe Werte, was darauf hindeutet, dass es Kernmerkmale der CTCF-Bindung erfasst, die in verschiedenen Geweben geteilt werden. Im Vergleich mit neun führenden Methoden, darunter frühere Deep-Learning- und BERT-basierte Modelle, zeigte TFBS-Finder eine höhere durchschnittliche Genauigkeit und lieferte verlässlichere Ranglisten von Bindungsstellen. Es lief zudem etwas schneller und verwendete weniger Speicher als das ähnlichste Vorgängermodell, was darauf hindeutet, dass bessere Leistung nicht unbedingt schwerere Rechnung erfordert.
Sehen, was das Modell gelernt hat
Komplexe KI-Systeme werden oft als „Black Boxes“ kritisiert. Die Forscher versuchten hier, diese Box zu öffnen, indem sie visualisierten, welche DNA-Positionen die Entscheidungen von TFBS-Finder am stärksten beeinflussten. Für zwei Transkriptionsfaktoren mit gut bekannten Bindungsmotiven, CEBPB und GATA3, erzeugten sie Wichtigkeitswerte entlang der Sequenz und gruppierten die stärksten Signale zu Konsensusmustern. Diese wiederhergestellten Motive stimmten eng mit Referenzmotiven aus etablierten Datenbanken überein, und die vorhergesagten Bindungsregionen überlappten mit unabhängig detektierten Motivinstanzen. Das legt nahe, dass TFBS-Finder nicht nur Beispiele auswendig lernt, sondern biologisch sinnvolle Regeln darüber erfasst hat, wie Transkriptionsfaktoren DNA erkennen.
Was das für Genetik und Medizin bedeutet
TFBS-Finder liefert eine genauere und besser interpretierbare Möglichkeit, die in unserer DNA eingebetteten Steuerungsmodule zu kartieren. Indem es punktgenau vorhersagt, wo Transkriptionsfaktoren wahrscheinlich binden, kann es Forschern helfen, Genregulationsnetzwerke zu kartieren, genetische Varianten zu priorisieren, die wichtige Kontrollstellen stören könnten, und gezieltere Experimente zu entwerfen. Obwohl die aktuelle Arbeit als künstliche Negative geschüttelte Sequenzen verwendet und sich nur auf DNA-Buchstaben konzentriert, planen die Autoren, strukturelle Informationen zur DNA-Form hinzuzufügen und realistischere Hintergrundsequenzen zu untersuchen. Wenn diese Modelle besser werden, könnten sie zu mächtigen Hilfsmitteln werden, um zu verstehen, wie Veränderungen in nichtkodierender DNA Entwicklung, Evolution und Krankheitsrisiken beeinflussen.
Zitation: Dutta, P., Ghosh, N. & Santoni, D. A DNABERT based deep learning framework for predicting transcription factor binding sites. Sci Rep 16, 7018 (2026). https://doi.org/10.1038/s41598-026-37483-1
Schlüsselwörter: Transkriptionsfaktor-Bindungsstellen, Deep Learning, DNABERT, Genregulation, Genomik