Clear Sky Science · de
GJB2 c.109G > A-Mutation aktiviert IFI27-vermittelte mitochondriale Apoptosewege und führt zu erblich bedingtem nicht-syndromischem Hörverlust
Warum winzige Ohrenzellen für die Zukunft von Kindern wichtig sind
Bei der Geburt vorhandener Hörverlust betrifft weltweit Millionen von Kindern und beeinflusst oft, wie sie Sprache lernen, in der Schule zurechtkommen und mit anderen in Kontakt treten. Einer der häufigsten genetischen Übeltäter ist das Gen GJB2, doch Ärzte haben bislang nicht vollständig verstanden, wie Veränderungen dieses Gens das Innenohr schädigen. Diese Studie nutzt Zebrafische und humanen Zelllinien, um die Kette von Ereignissen von einer einzigen DNA-Veränderung in GJB2 bis zum Absterben empfindlicher schallwahrnehmender Zellen nachzuzeichnen, und macht ein neues Molekül, IFI27, als möglichen Ansatzpunkt für künftige Therapien ausfindig.
Eine häufige Genveränderung hinter stillem Kind- gehör
Die Forschenden begannen mit dem Screening von Blutproben von 1.199 Kindern mit Verdacht auf erblichen Hörverlust in der Provinz Fujian, China. Sie konzentrierten sich auf mehrere bekannte Taubheitsgene und stellten fest, dass Veränderungen in GJB2 dominierten und 85 % aller detektierten Mutationen ausmachten. Unter diesen war eine spezifische Veränderung, genannt c.109G>A (auch bekannt als p.Val37Ile), die häufigste. Diese Variante ist in der Allgemeinbevölkerung relativ verbreitet, bei Personen mit Hörverlust jedoch stark angereichert, was darauf hinweist, dass sie eine bedeutende Rolle beim nicht-syndromischen Hörverlust spielt — also Hörproblemen, die ohne zusätzliche medizinische Auffälligkeiten auftreten.
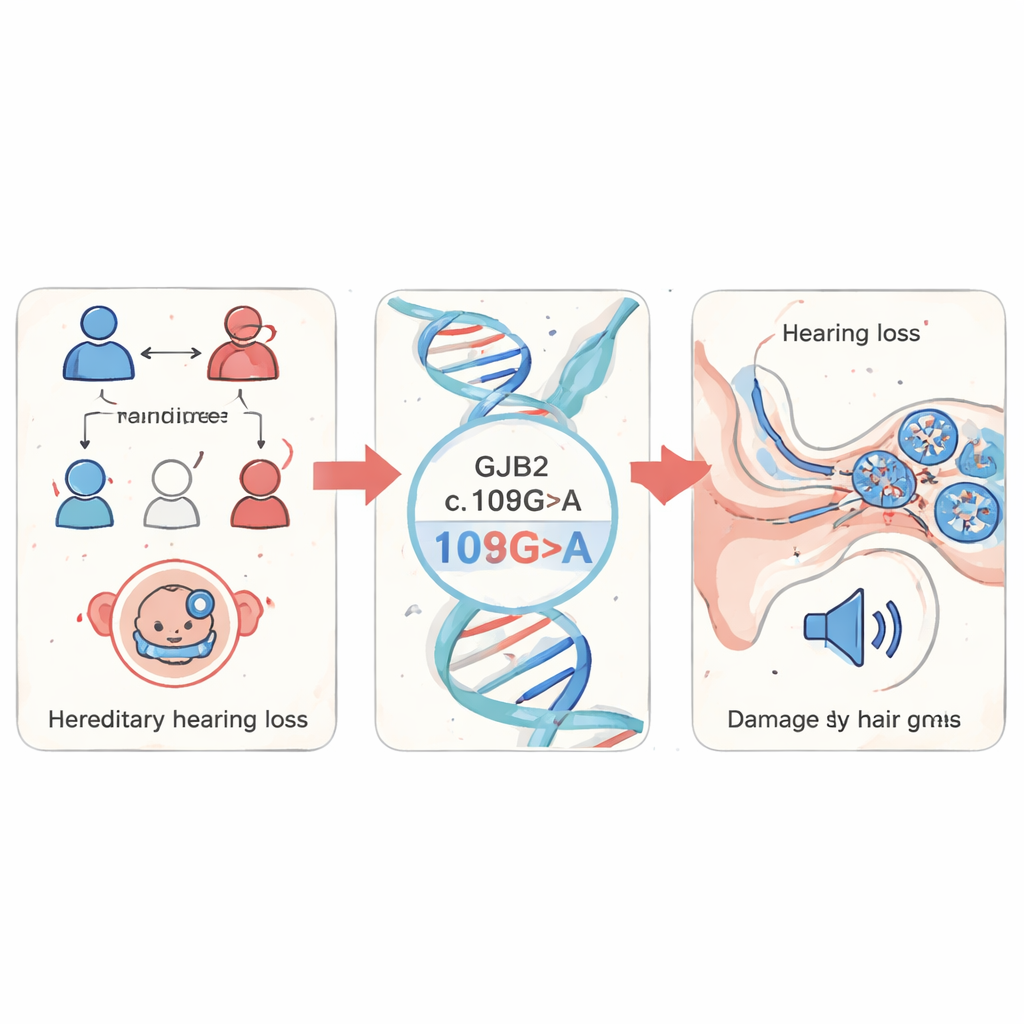
Dem Schaden in einem transparenten Fisch folgen
Um zu sehen, was diese Mutation in einem lebenden Organismus bewirkt, griff das Team auf den Zebrafisch zurück, einen kleinen Süßwasserfisch, dessen Embryonen durchsichtig sind und der viele Gene und Ohrstrukturen mit Menschen teilt. Sie erzeugten Zebrafisch-Embryonen, die entweder das normale menschliche GJB2 oder die c.109G>A-Mutante produzierten, und setzten zusätzlich einen „Knockdown“-Ansatz ein, um das eigene gjb2 der Fische zu reduzieren. Embryonen mit der mutierten oder reduzierten Version des Gens zeigten verzögertes Wachstum, gekrümmte Schwänze und Schwellungen um das Herz — Anzeichen für fehlgeleitete Entwicklung. Am wichtigsten war, dass ihre Innenohren deutlich abnormal waren: Schlüsselstrukturen, die Otolithen, waren kleiner und weiter auseinander, und der flüssigkeitsgefüllte Kochlearbereich war verkleinert. Als die Forschenden zusätzlich normales GJB2 zusammen mit der Mutante hinzugaben, verbesserten sich viele dieser strukturellen Probleme, was zeigte, dass die Mutation selbst die Defekte verursachte.
Von fehlerhaften Ohren zu schlechterem Hörverhalten
Da das Hören auf winzige „Haarsinneszellen“ angewiesen ist, die Schallvibrationen in Nervenimpulse umwandeln, färbte das Team diese Zellen in den Zebrafischen an. Fische mit der GJB2-Mutation oder dem Knockdown hatten deutlich weniger Haarsinneszellen sowohl im Innenohr als auch entlang der Körperoberfläche, wo Zebrafische ebenfalls Wasserbewegungen wahrnehmen. Anschließend testeten die Forschenden, wie gut die Fische auf Schall reagierten. Mit einem automatisierten Tracking-System maßen sie, wie weit und wie schnell 5 Tage alte Larven schwammen, wenn sie kurzen Schallstößen ausgesetzt wurden. Normale und Wildtyp-GJB2-Fische reagierten mit mehr und schnellerer Schwimmbewegung, während mutierte und Knockdown-Fische ihr Verhalten kaum veränderten — ein Hinweis auf eingeschränktes Hören. Auch hier stellte das zusätzliche Einbringen von normalem GJB2 teilweise sowohl die Anzahl der Haarsinneszellen als auch die schallgetriebene Bewegung wieder her.
Ein tödlicher Weg in den Kraftwerken der Zelle
Um zu verstehen, was sich in den Zellen abspielte, verwendeten die Wissenschaftler RNA-Sequenzierung, um die Genaktivität zwischen normalen Zebrafischen und solchen mit reduziertem gjb2 zu vergleichen. Eine Gruppe von Genen, die mit dem „mitochondrialen Apoptoseweg“ verbunden sind — einem Selbstzerstörungsprogramm, das in den Energiezentralen der Zelle stattfindet — war stark aktiviert. Besonders mehrere Mitglieder der IFI27-Familie stachen hervor, ebenso wie bekannte Todesakteure wie Bax, Cytochrom c, Apaf1 und Caspasen. Nachfolgende Experimente in humanen HEK293-Zellen bestätigten dieses Muster: Zellen mit der mutierten GJB2 produzierten mehr reaktive Sauerstoffspezies (ROS, eine Form von oxidativem Stress), setzten mehr Cytochrom c aus den Mitochondrien frei und schalteten Apoptose-Proteine an, was zu vermehrtem Zelltod führte. Als die Forschenden IFI27 in Zellen mit der Mutante stilllegten, sanken die ROS-Werte, die Todessignale wurden abgeschwächt und weniger Zellen durchliefen Apoptose.
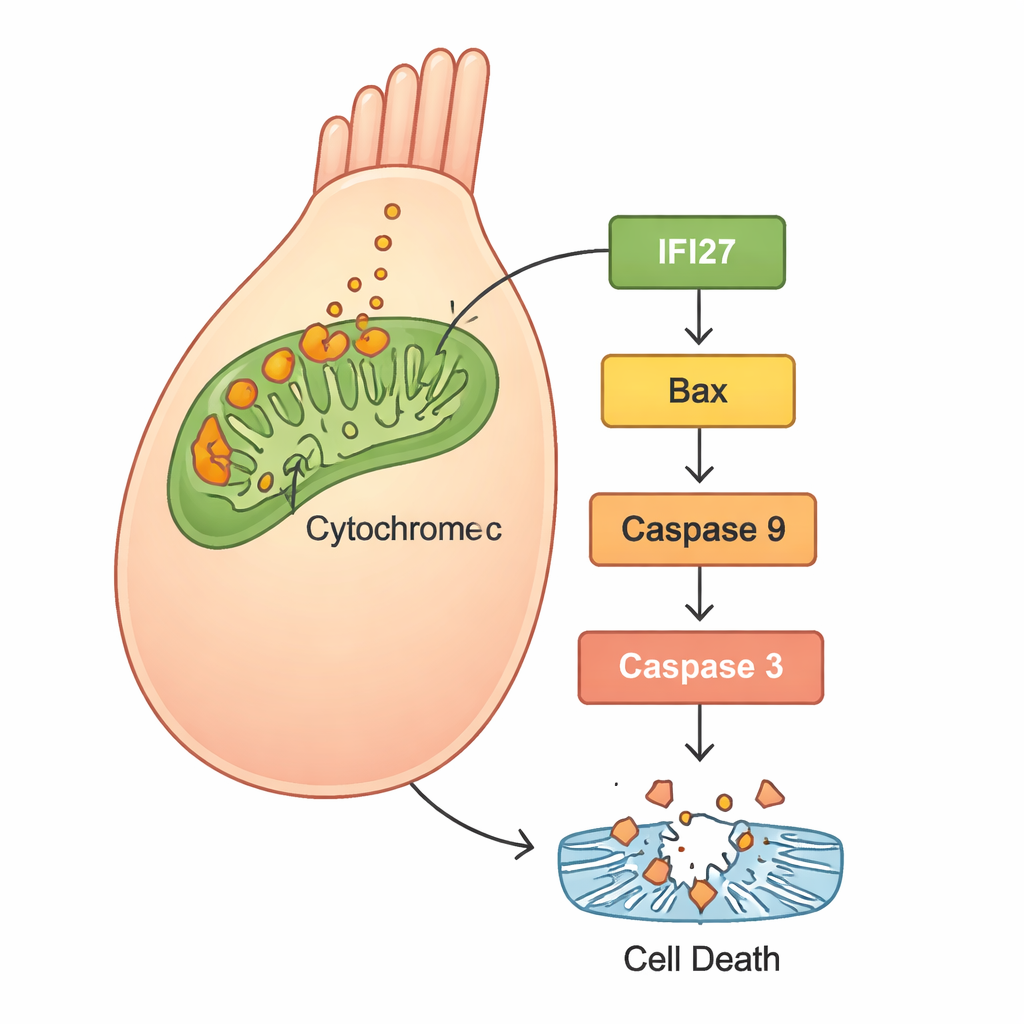
Was das für künftige Behandlungen bedeutet
Insgesamt deuten die Befunde auf eine klare Geschichte hin: Die GJB2 c.109G>A-Mutation stört die normale Entwicklung und Funktion des Innenohrs, nicht nur durch veränderte Zellkommunikation, sondern auch durch Auslösung von Stress in den Mitochondrien. Dieser Stress erhöht IFI27 und verwandte Gene, führt zur Freisetzung von Cytochrom c und aktiviert eine Kaskade von Proteinen, die Haarsinneszellen in Richtung programmierter Zelltodeswege treiben. Da Haarsinneszellen beim Menschen nur schlecht nachwachsen, führt ihr Verlust zu dauerhaften Hördefiziten. Indem die Studie zeigt, dass das Herunterregeln von IFI27 diese zerstörerische Kaskade in menschlichen Zellen abschwächen kann, hebt sie IFI27 als vielversprechendes Ziel für Medikamente oder gentherapeutische Ansätze hervor. Solche Behandlungen sind zwar noch Zukunftsmusik — und müssten vermutlich sehr früh im Leben verabreicht werden — doch liefert diese Arbeit eine konkrete molekulare Roadmap, um eine einst rätselhafte Genmutation in eine potenziell vermeidbare Ursache kindlicher Taubheit zu verwandeln.
Zitation: Chen, Y., Zhao, P., Lin, Q. et al. GJB2 c.109G > A mutation activating IFI27-mediated mitochondrial apoptosis pathway leading to hereditary non-syndromic hearing loss. Sci Rep 16, 6240 (2026). https://doi.org/10.1038/s41598-026-37393-2
Schlüsselwörter: genetisch bedingter Hörverlust, GJB2-Mutation, Zebrafischmodell, mitochondriale Apoptose, IFI27