Clear Sky Science · de
Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data
Warum das für die Genforschung wichtig ist
Moderne Genexpressions‑Tests können in einer einzelnen Patientenprobe Zehntausende von Genen messen. Diese Datenflut verspricht frühere Krebsdiagnosen und bessere Therapieentscheidungen, erzeugt aber auch ein Problem: die meisten dieser Gene sind verrauscht, redundant oder beziehen sich hauptsächlich auf häufige Fälle, nicht die seltenen und gefährlichen. Diese Arbeit stellt eine neue Methode vor, um massive Genexpressionsdatensätze zu durchsieben, sodass Computer zuverlässig Patienten in einer kleinen, schwer zu entdeckenden Minderheitsgruppe anhand nur einer winzigen, sorgfältig ausgewählten Genmenge identifizieren können.
Die Herausforderung: zu viele, zu ähnliche Gene
Microarray‑Experimente verfolgen oft tausende Genaktivitätswerte, haben aber nur wenige hundert Proben. Meist überwiegt eine Klasse (etwa ein häufiger Krebsuntertyp) die andere deutlich, sodass die Daten stark unausgeglichen sind. In diesem Umfeld verhalten sich viele Gene sehr ähnlich, und die Muster für Mehrheits‑ und Minderheitspatienten können überlappen. Standard‑Lernverfahren neigen dazu, sich an der Mehrheitsklasse auszurichten und von redundanten Genen verwirrt zu werden, was zu Overfitting und schlechter Erkennung seltener Subtypen führt. Traditionelle Verfahren zur Dimensionsreduktion opfern entweder Interpretierbarkeit, indem sie neue gemischte Merkmale erzeugen, oder wählen Gene aus, ohne genau zu prüfen, wie sehr sie einem Klassifikator bei der Erkennung der Minderheitsfälle helfen.
Ein neuer Fahrplan für intelligentere Genauswahl
Die Autorinnen und Autoren stellen AFCG‑SFE vor, ein adaptives Merkmalsauswahlmodell, das speziell für hochdimensionale, unausgeglichene Genexpressionsdaten entwickelt wurde. Die Methode beginnt mit einer einfachen "Single‑Agent"‑Suche, die Gene ein‑ oder ausschaltet und testet, wie gut sie die Klassifikation unterstützen, und ergänzt dies durch mehrere datengesteuerte Schritte. Zuerst gruppiert sie Gene nach ähnlichem Verhalten und erlaubt, dass Gene mehreren Gruppen angehören, um der biologischen Realität Rechnung zu tragen, dass ein Gen an mehreren Signalwegen beteiligt sein kann. Innerhalb jeder Gruppe werden Gene nach ihrer Informationsstärke bezüglich des Krankheitslabels gerankt und nur wenige zentrale Vertreter behalten, wodurch Redundanz bereits vor der eigentlichen Suche stark reduziert wird. 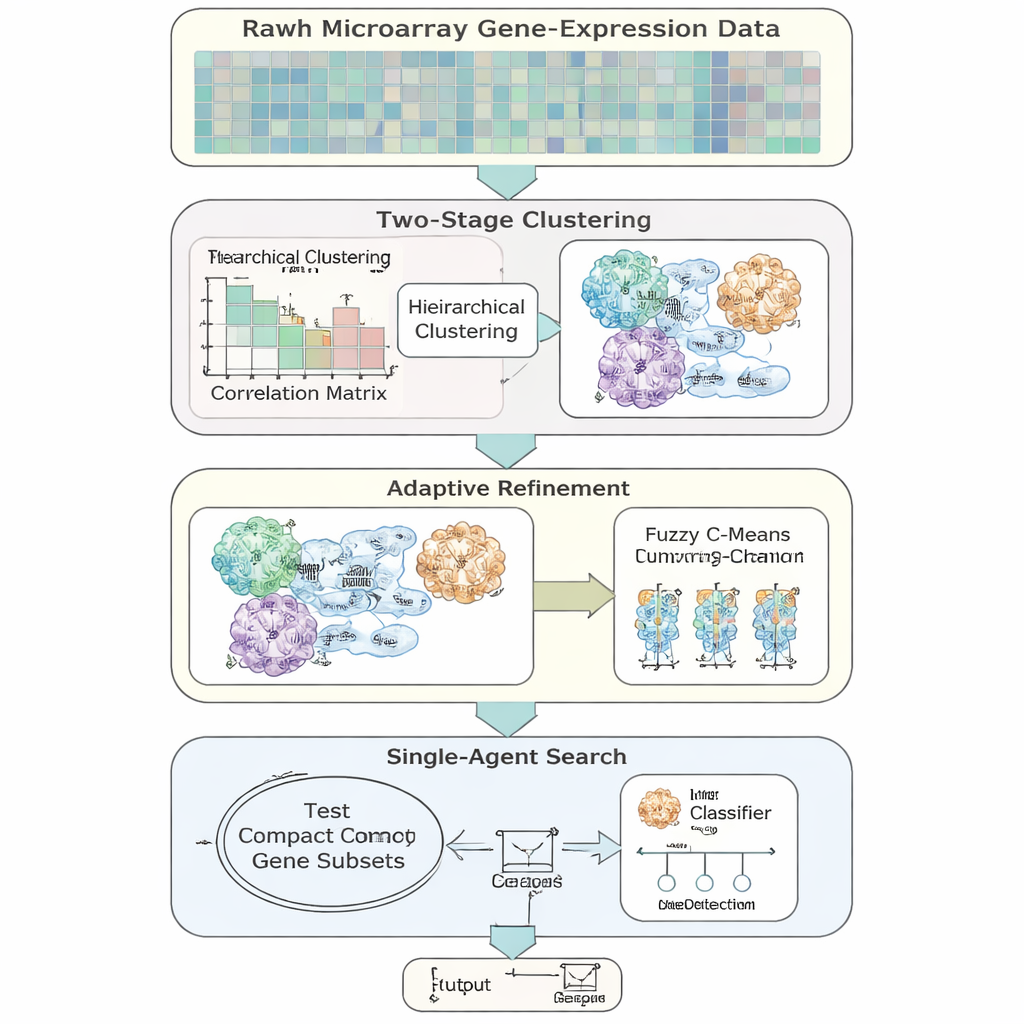
Den Computer für seltene Patientinnen und Patienten sensibilisieren
Statt sich auf einfache Genauigkeit zu konzentrieren, verwendet AFCG‑SFE eine Fitness‑Metrik, die Kennzahlen betont, die für schiefe Daten geeignet sind, einschließlich des Gleichgewichts zwischen korrekter Identifikation von Minderheits‑ und Mehrheitsfällen und der Leistung über alle Entscheidungs‑Schwellen hinweg. Die Fitnessfunktion enthält zudem Straftermine für die Auswahl zu vieler Gene oder vieler Gene aus demselben Cluster sowie eine Belohnung für Gene, die eine starke Abhängigkeit vom Krankheitslabel tragen. Wichtig ist, dass die Stärke dieser Strafen und Belohnungen automatisch aus Eigenschaften des Datensatzes abgeleitet wird — etwa aus der Anzahl der Gene pro Probe und dem Ausmaß der Klassenüberlappung — statt durch manuelle Abstimmung. Das macht die Methode robuster und leichter zwischen Studien übertragbar.
Anpassung an die Schwierigkeit des Problems
Eine Kernidee ist, dass der Algorithmus nicht immer auf die kleinstmögliche Genmenge zielen sollte. Wenn die beiden Klassen schwer zu trennen oder stark überlappt sind, erhöht die Methode automatisch eine Untergrenze für die Anzahl der zu behaltenden Gene, um sicherzustellen, dass seltene, aber wichtige Signale nicht verworfen werden. Im Verlauf der Suche verschärft AFCG‑SFE schrittweise eine pro‑Cluster Obergrenze dafür, wie viele Gene aus jeder Gruppe überleben dürfen, während diese Mindestanzahl respektiert wird. Das Ergebnis ist ein kompaktes, vielfältiges Panel von Genen, das die Struktur der Daten erfasst, ohne von einem einzigen redundanten Muster dominiert zu werden. 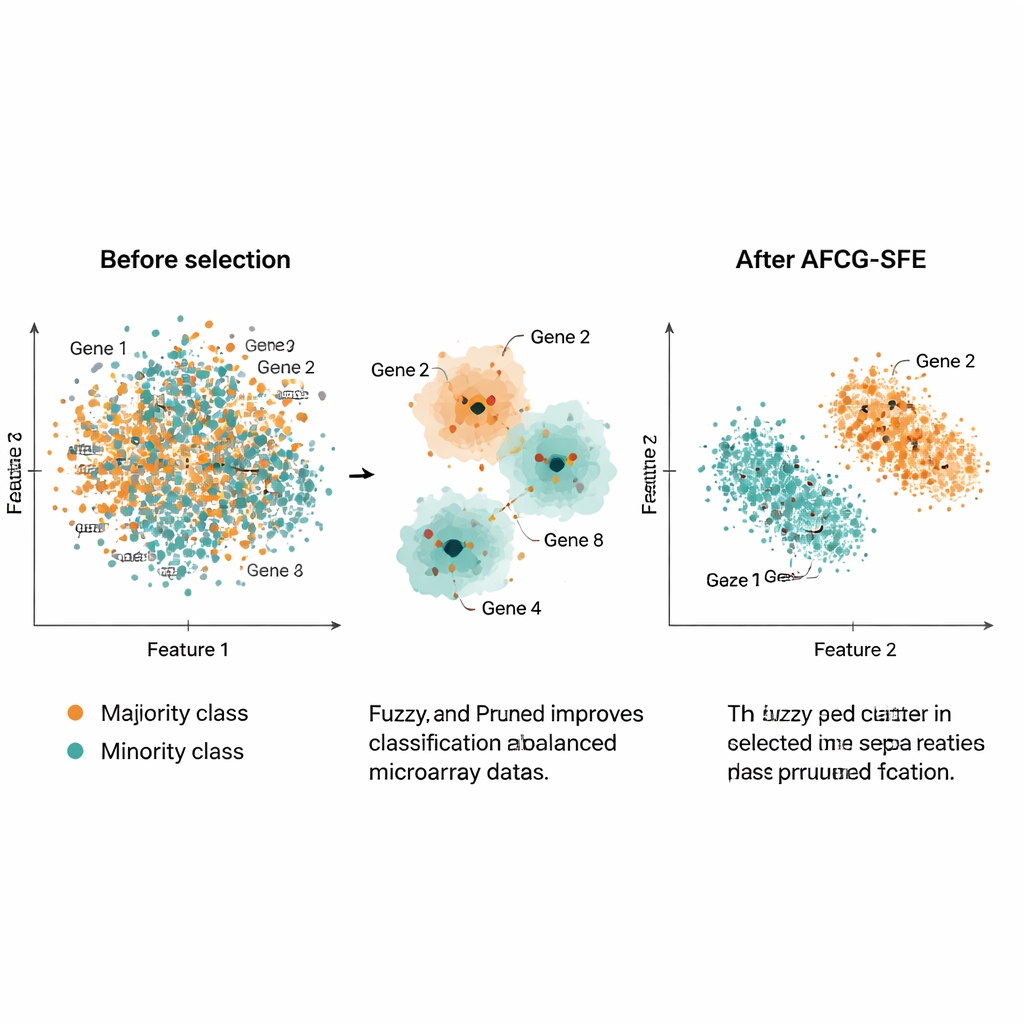
Was die Experimente zeigen
Die Autorinnen und Autoren testeten AFCG‑SFE an 20 öffentlichen Krebs‑Microarray‑Datensätzen, jeweils mit tausenden Genen, aber nur etwa 100–200 Proben und starker Klassenungleichheit. Sie verglichen ihre Methode mit mehreren evolutionären Suchverfahren als Baselines, einfachen Filtern und eingebetteten Ansätzen, die Merkmalsauswahl in den Klassifikator integrieren. Über eine Reihe von Metriken hinweg — einschließlich F‑Metrik, balancierter Genauigkeit, Fläche unter der ROC‑Kurve und einem Maß für Overfitting — war AFCG‑SFE auf allen Datensätzen führend oder geteilter Spitzenreiter. Typischerweise wählte es weniger als 25 Gene aus (oft nur 6–8) und entfernte mehr als 99 % der ursprünglichen Merkmale, während es die Klassifikationsleistung verbesserte oder beibehielt. Außerdem verringerte es einen Komplexitätsindex, der erfasst, wie stark die Klassen im Merkmalsraum überlappen, was auf eine klarere Trennung nach der Auswahl hindeutet.
Fazit für Nicht‑Expertinnen und Nicht‑Experten
Praktisch bietet diese Arbeit eine Möglichkeit, riesige, verrauschte Genexpressionsprofile auf sehr kleine, informative Genmengen zu reduzieren, die dennoch Computer in die Lage versetzen, seltene Patientengruppen zuverlässig zu erkennen. Durch intelligentes Gruppieren ähnlicher Gene, das Belohnen solcher, die das Krankheitsgeschehen wirklich abbilden, und den expliziten Schutz vor einer Verzerrung zugunsten der Mehrheitsklasse liefert AFCG‑SFE sowohl bessere Vorhersagen als auch deutlich einfachere Genpanels. Diese Kombination kann Forschenden helfen, potenzielle Biomarker zu identifizieren, interpretierbarere diagnostische Tests zu entwerfen und letztlich zu verbessern, wie Werkzeuge der Präzisionsmedizin mit realen, unvollkommenen biologischen Daten arbeiten.
Zitation: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
Schlüsselwörter: Genexpressionsdaten, Merkmalsauswahl, unausgeglichene Daten, Microarray, Krebssubtypen