Clear Sky Science · de
rhinotypeR ermöglicht reproduzierbare Bestimmung von Rhinovirus‑Genotypen aus VP4/2‑Sequenzen
Warum winzige Erkältungsviren weiterhin wichtig sind
Die meisten von uns betrachten die gewöhnliche Erkältung eher als lästige Erscheinung denn als ernsthafte Bedrohung. Dennoch stehen viele der Erkältungsviren — die humanen Rhinoviren — auch im Zusammenhang mit schweren Lungeninfektionen, Asthmaanfällen und Verschlechterungen chronischer Lungenerkrankungen. Um nachzuverfolgen, wie sich diese Viren entwickeln und verbreiten, müssen Forscher sie in präzise genetische „Typen“ einteilen, ähnlich dem Zuweisen von Barcodes zu Produkten. Dieser Artikel stellt rhinotypeR vor, ein kostenloses, quelloffenes Softwarepaket, das diese genetische Kennzeichnung genauer, konsistenter und leichter reproduzierbar macht und so den Gesundheitsämtern hilft, ein oft übersehenes Familienmitglied der Atemwegsviren klarer zu beobachten.
Die verborgene Vielfalt der gewöhnlichen Erkältungen
Humane Rhinoviren sind außerordentlich verbreitet und finden sich in bis zu 60 % der Proben von Menschen mit akuter Atemwegserkrankung. Sie sind keineswegs ein einzelnes Virus, sondern in drei Hauptgruppen unterteilt, A, B und C, und umfassen mindestens 169 anerkannte genetische Typen. Verschiedene Typen verhalten sich unterschiedlich: Einige werden häufiger mit schweren Infektionen bei Kindern und mit Asthmaexazerbationen in Verbindung gebracht, andere treten seltener bei schweren Erkrankungen auf. Weil diese Typen unabhängig voneinander evolvieren und unterschiedliche Oberflächenmerkmale tragen, benötigen Wissenschaftler verlässliche Methoden, um sie zu unterscheiden, wenn sie Ausbrüche in Schulen, Haushalten und Gemeinden verfolgen wollen.
Von verstreuten Werkzeugen zu einem klaren Ablauf
Bisher war die Bestimmung eines Rhinovirus‑Typs aus dessen genetischem Code ein Flickwerk. Forscher konzentrierten sich meist auf einen kurzen Abschnitt im Virusgenom, die VP4/2‑Region, richteten ihn an bekannten Referenzstämmen aus, bestimmten die Sequenzdistanzen und wandten dann Cut‑off‑Werte an, um jedem Proben die passende Typenbezeichnung zuzuweisen. Diese Schritte wurden jedoch mit einer Mischung aus verschiedenen Programmen, manuellen Korrekturen und persönlichem Ermessen durchgeführt. Das erschwerte den Vergleich oder die Reproduzierbarkeit unterschiedlicher Studien, selbst bei ähnlichen Daten. rhinotypeR wurde speziell entwickelt, um diesen mehrstufigen, fehleranfälligen Prozess in einen einzigen, skriptbaren Workflow zu verwandeln, den jeder ausführen und weitergeben kann.
Was die neue Software praktisch leistet
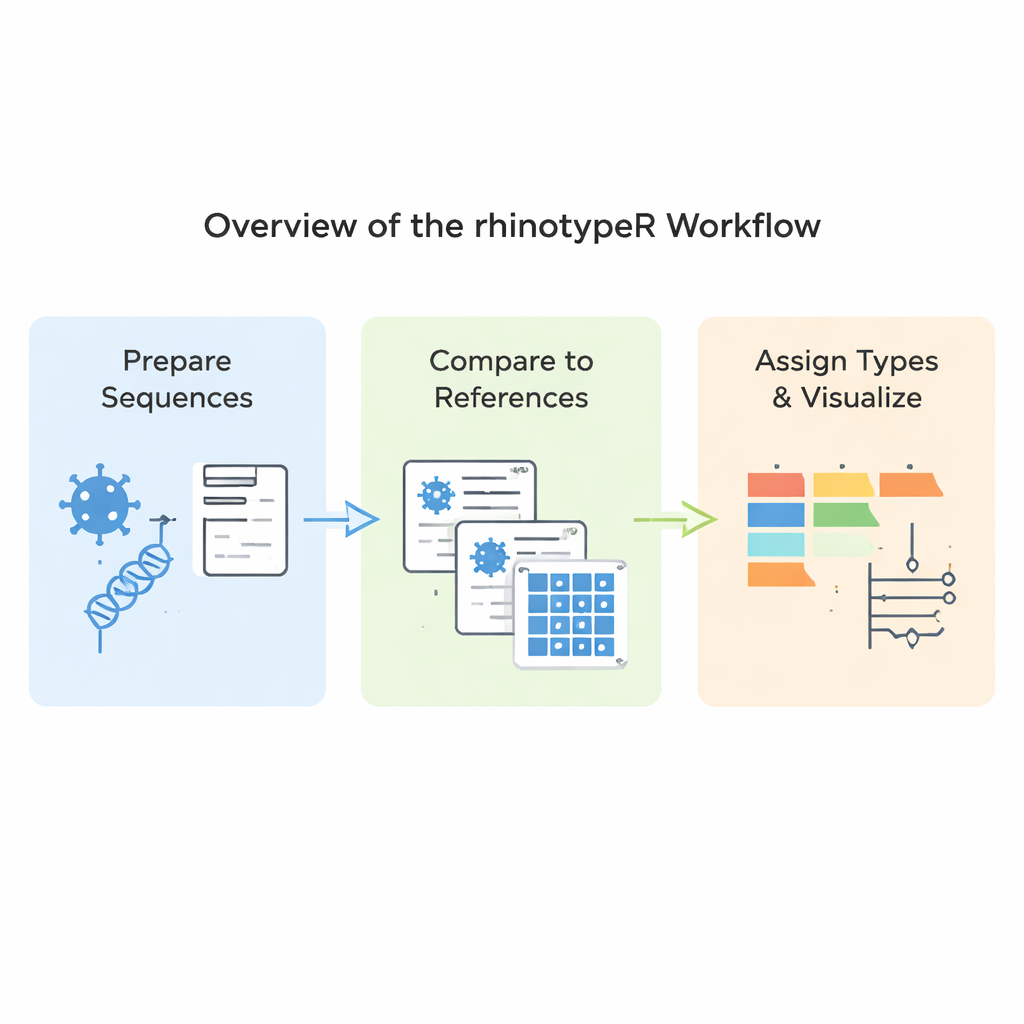
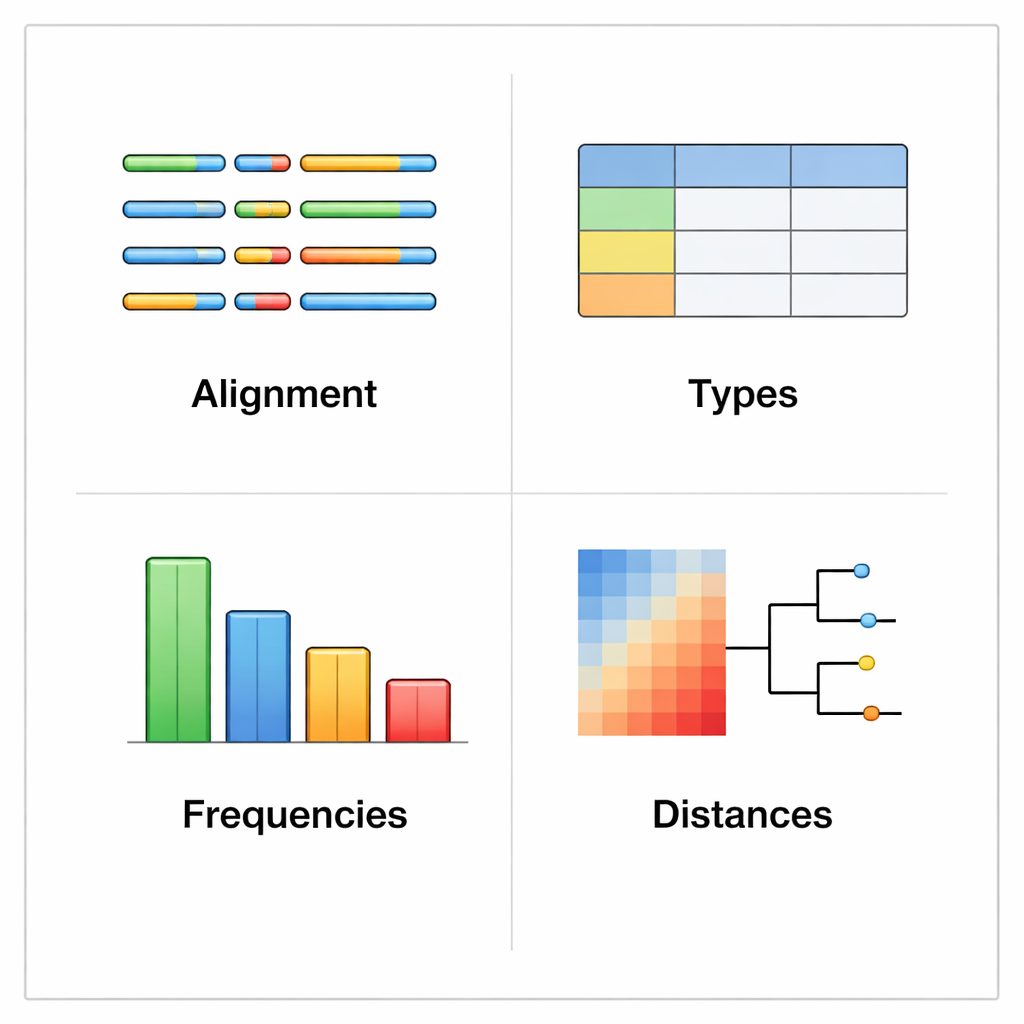
rhinotypeR läuft in der weit verbreiteten R‑ und Bioconductor‑Umgebung für Datenanalyse. Es nimmt eine Sammlung von Rhinovirus‑VP4/2‑Sequenzen auf und führt sie durch drei Hauptphasen: Vorbereitung und Ausrichtung der Sequenzen, Berechnung der Abstände zu einer kuratierten Referenztypliste und anschließende Zuordnung jeder Probe zum nächstgelegenen bekannten Typ oder Kennzeichnung als „unassigned“, falls sie zu unterschiedlich ist. Dasselbe Werkzeug kann visuelle Ausgaben erzeugen, darunter farbkodierte Karten genetischer Unterschiede, einfache Stammbaumdarstellungen und Diagramme, die die Häufigkeit der Typen in einem Datensatz zeigen. Nutzer können ihre Daten mit externen Programmen ausrichten, wenn sie das bevorzugen, oder rhinotypeR den gesamten Prozess innerhalb von R durchführen lassen, um maximale Reproduzierbarkeit zu erreichen.
Wie das Werkzeug geprüft wurde
Um zu überprüfen, dass rhinotypeR vertrauenswürdige Ergebnisse liefert, verglichen die Autorinnen und Autoren dessen Distanzmessungen mit denen von zwei etablierten Programmen, ape und MEGA X, unter Verwendung derselben Eingabedateien und Modelle. Die Ergebnisse stimmten nahezu perfekt überein; winzige Abweichungen waren auf übliche Rundungsfehler in der Computerarithmetik zurückzuführen, nicht auf methodische Unterschiede. Anschließend setzte das Team rhinotypeR auf einer großen Sammlung von mehr als 2.300 Rhinovirus‑Sequenzen aus mehreren früheren Studien ein, die über 90 % der bekannten Typen abdeckten. In etwa vier von fünf Fällen stimmte das neue Werkzeug genau mit den früheren Typenbezeichnungen überein. Die meisten Unstimmigkeiten traten genau an den zuvor festgelegten Cut‑off‑Punkten auf, also dort, wo Grenzfälle zu erwarten sind. Wichtig ist, dass Proben, die nicht zuverlässig einem bekannten Typ zugeordnet werden konnten, keine Hinweise auf schlechte Probenqualität oder niedrige Viruslast zeigten, was nahelegt, dass sie echte virale Vielfalt widerspiegeln könnten.
Warum das für die öffentliche Gesundheit wichtig ist
Für Nicht‑Spezialisten ist die Kernbotschaft: rhinotypeR erfindet die Klassifizierung von Erkältungsviren nicht neu, sondern macht den Prozess transparenter, nachvollziehbarer und leichter reproduzierbar. Indem es Ausrichtung, Distanzberechnungen und Typzuordnung in einem offenen Paket bündelt — zusammen mit klaren visuellen Zusammenfassungen — hilft es Forschern und Überwachungsprogrammen, Tausende von Proben konsistent zu verarbeiten. Diese Konsistenz verbessert unsere Fähigkeit, Studien aus verschiedenen Regionen und Zeiträumen zu vergleichen, ungewöhnliche oder neu auftretende Viruslinien frühzeitig zu erkennen und genetische Muster mit realen Krankheitsgeschehen zu verknüpfen. Langfristig stärken Werkzeuge wie rhinotypeR die routinemäßige Überwachung scheinbar gewöhnlicher Erkältungen, die bei vielen Menschen schwere Erkrankungen auslösen können.
Zitation: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Schlüsselwörter: Rhinovirus‑Genotypisierung, molekulare Überwachung, VP4/2‑Sequenzierung, Bioinformatik‑Werkzeuge, Atemwegsviren